Department of Biostatistics, Harvard University, Boston, MA, USA.
PLoS One. 2013;8(1):e48495. doi: 10.1371/journal.pone.0048495. Epub 2013 Jan 15.
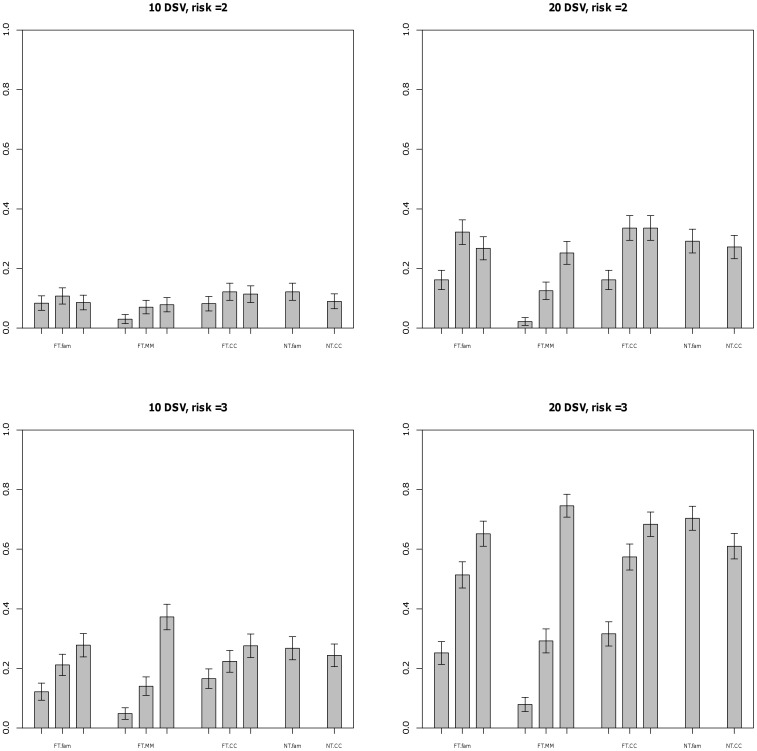
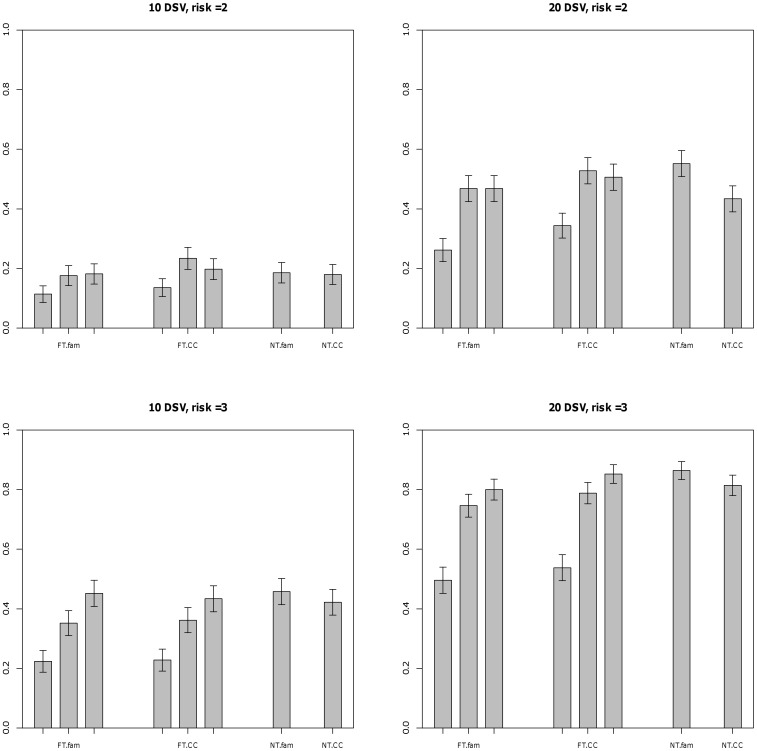
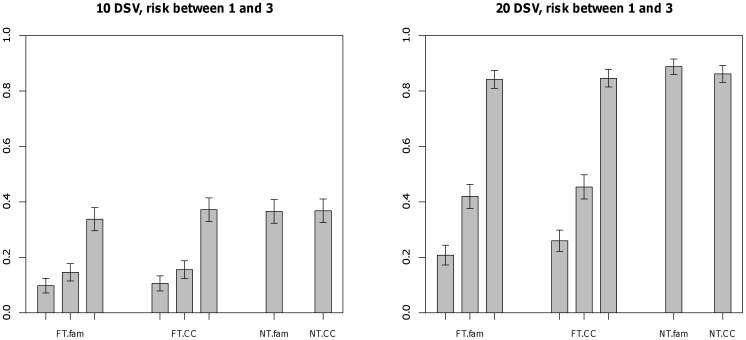
Genome-wide association studies have been able to identify disease associations with many common variants; however most of the estimated genetic contribution explained by these variants appears to be very modest. Rare variants are thought to have larger effect sizes compared to common SNPs but effects of rare variants cannot be tested in the GWAS setting. Here we propose a novel method to test for association of rare variants obtained by sequencing in family-based samples by collapsing the standard family-based association test (FBAT) statistic over a region of interest. We also propose a suitable weighting scheme so that low frequency SNPs that may be enriched in functional variants can be upweighted compared to common variants. Using simulations we show that the family-based methods perform at par with the population-based methods under no population stratification. By construction, family-based tests are completely robust to population stratification; we show that our proposed methods remain valid even when population stratification is present.
全基因组关联研究已经能够识别许多常见变异与疾病的关联;然而,这些变异所解释的遗传贡献估计大部分都非常有限。与常见的 SNP 相比,稀有变异被认为具有更大的效应大小,但在 GWAS 环境中无法检测到稀有变异的效应。在这里,我们提出了一种新的方法,通过对基于家族的样本进行测序来检测罕见变异的关联,通过在感兴趣的区域上折叠标准的基于家族的关联测试(FBAT)统计量。我们还提出了一个合适的加权方案,以便可以对功能变异中可能富集的低频 SNP 进行加权,与常见变异相比。通过模拟,我们表明在没有群体分层的情况下,基于家族的方法与基于人群的方法表现相当。通过构造,基于家族的测试完全不受群体分层的影响;我们表明,即使存在群体分层,我们提出的方法仍然有效。