Department of Pathology, Memorial Sloan-Kettering Cancer Center, New York, NY 10021, USA.
Am J Surg Pathol. 2013 Mar;37(3):385-92. doi: 10.1097/PAS.0b013e31826c1761.
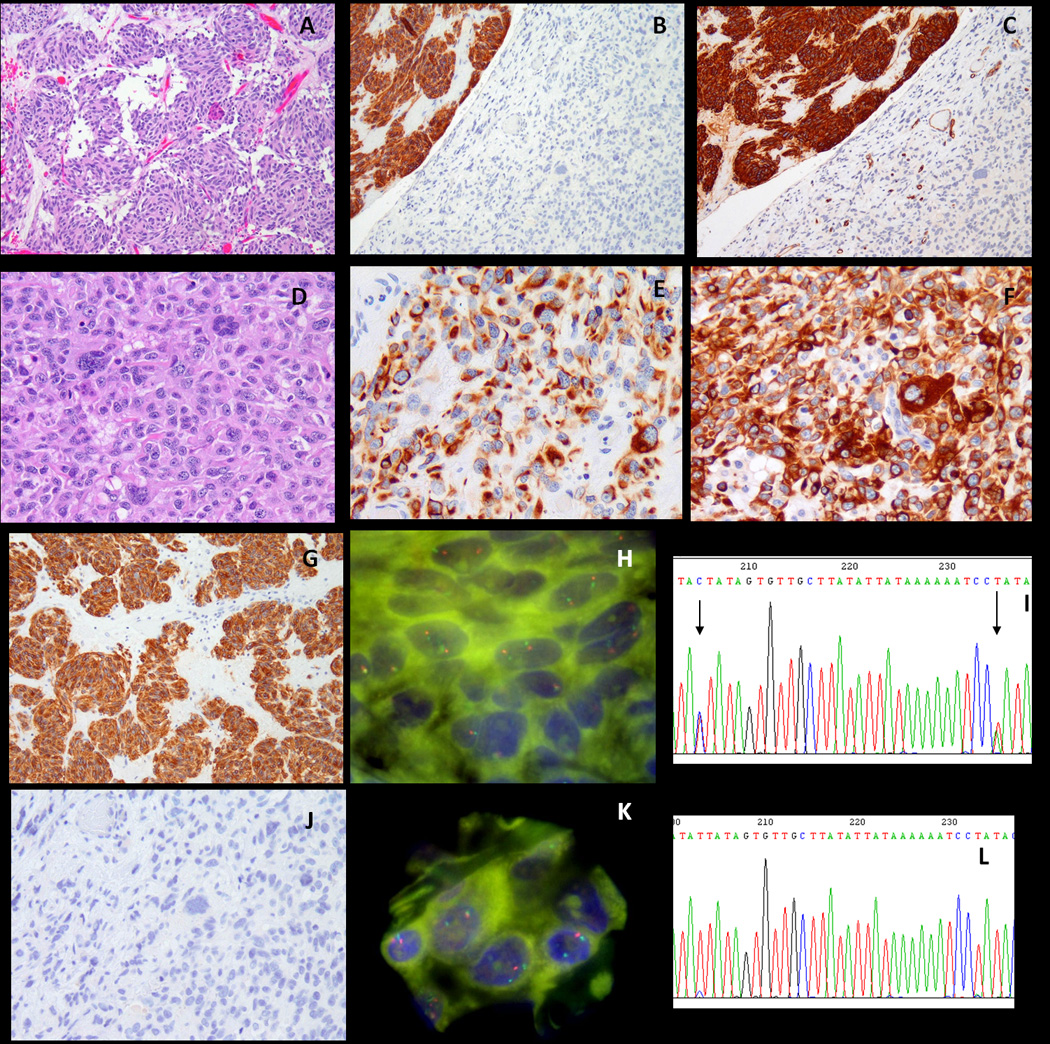
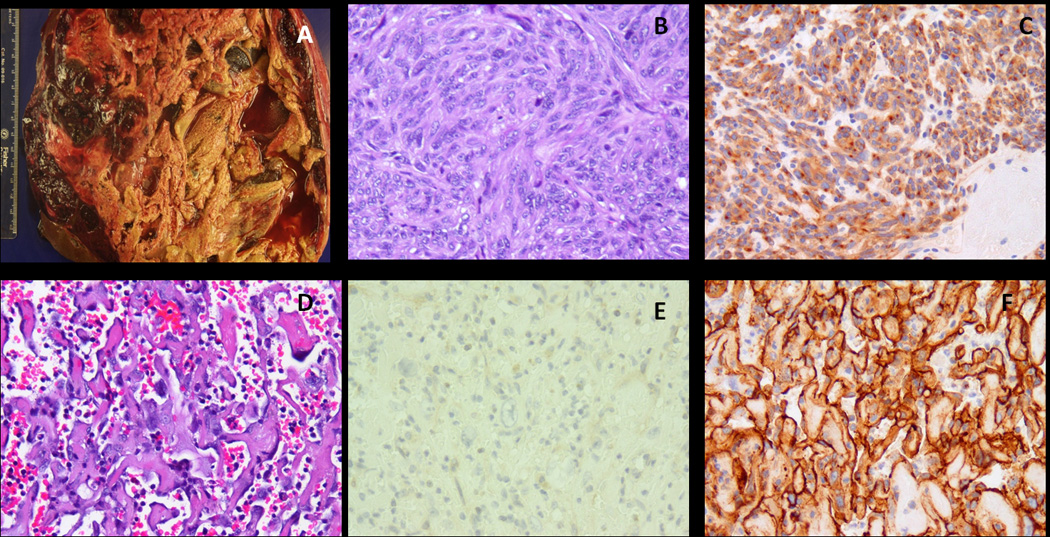
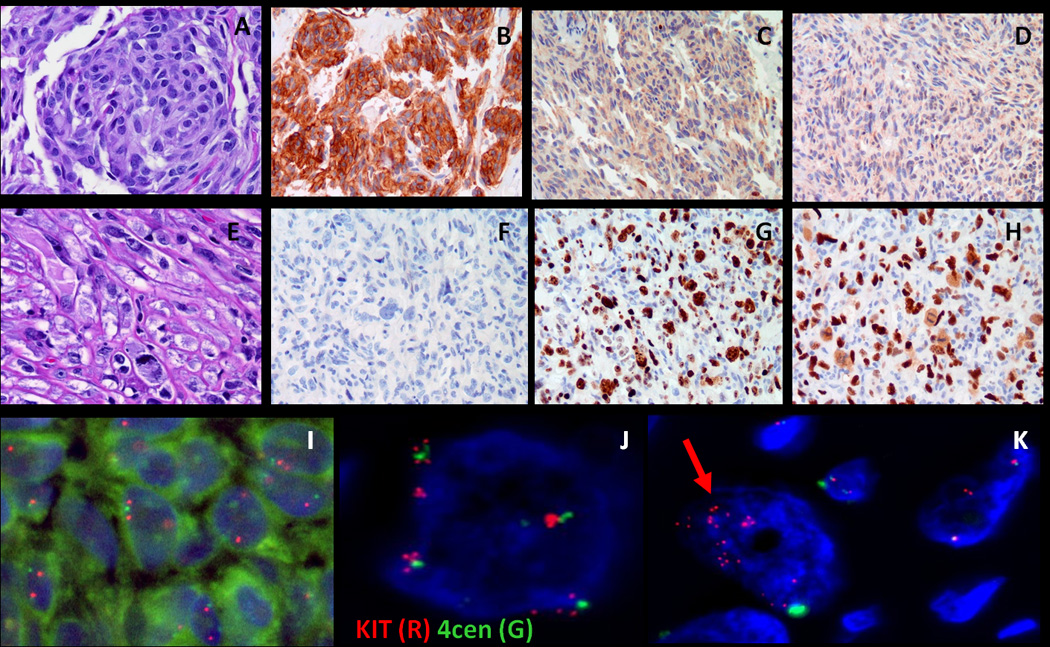
Most gastrointestinal stromal tumors (GISTs) can be recognized by their monotonous cytologic features and overexpression of KIT oncoprotein. Altered morphology and loss of CD117 reactivity has been described previously after chronic imatinib treatment; however, this phenomenon has not been reported in imatinib-naive tumors. Eight patients with abrupt transition from a classic CD117-positive spindle cell GIST to an anaplastic CD117-negative tumor were investigated for underlying molecular mechanisms of tumor progression. Pathologic and molecular analysis was performed on each of the 2 components. Genomic DNA polymerase chain reaction for KIT, PDGFRA, BRAF, and KRAS hot spot mutations and fluorescence in situ hybridization for detecting KIT gene copy number alterations were performed. TP53 mutational analysis was performed in 5 cases. There were 7 men and 1 woman, with an age range of 23 to 65 years. Five of the primary tumors were located in the stomach, and 1 case each originated in the small bowel, colon, and rectum. In 3 patients, the dedifferentiated component occurred in the setting of imatinib resistance, whereas the remaining 5 occurred de novo. The dedifferentiated component had an anaplastic appearance, including 1 angiosarcomatous phenotype, with high mitotic activity and necrosis, and showed complete loss of CD117 (8/8) and CD34 (5/8) expression and de novo expression of either cytokeratin (4/8) or desmin (1/8). There was no difference in the KIT genotype between the 2 components. However, 2 imatinib-resistant tumors showed coexistence of KIT exon 11 and exon 13 mutations. Fluorescence in situ hybridization showed loss of 1 KIT gene in 3 cases and low-level amplification of KIT in 2 other cases in the CD117-negative component, compared with the CD117-positive area. TP53 mutation was identified in 1/5 cases tested, being present in both components. In summary, dedifferentiation in GIST may occur either de novo or after chronic imatinib exposure and can represent a diagnostic pitfall. This phenomenon is not related to additional KIT mutations, but might be secondary to genetic instability, either represented by loss of heterozygosity or low level of KIT amplification.
大多数胃肠道间质瘤(GIST)可以通过其单调的细胞学特征和 KIT 癌蛋白的过表达来识别。先前已经描述了在慢性伊马替尼治疗后形态改变和 CD117 反应性丧失;然而,这种现象在伊马替尼初治肿瘤中尚未报道。8 例经典 CD117 阳性梭形细胞 GIST 突然转变为间变性 CD117 阴性肿瘤的患者,研究了肿瘤进展的潜在分子机制。对每个 2 个成分进行了病理和分子分析。进行了 KIT、PDGFRA、BRAF 和 KRAS 热点突变的基因组 DNA 聚合酶链反应和 KIT 基因拷贝数改变的荧光原位杂交检测。在 5 例中进行了 TP53 突变分析。患者为 7 男 1 女,年龄 23 至 65 岁。5 例原发肿瘤位于胃,1 例分别起源于小肠、结肠和直肠。在 3 例患者中,去分化成分发生在伊马替尼耐药的情况下,而其余 5 例则是初发的。去分化成分表现出间变性外观,包括 1 例血管肉瘤表型,具有高有丝分裂活性和坏死,并完全丧失 CD117(8/8)和 CD34(5/8)表达,并新表达细胞角蛋白(4/8)或结蛋白(1/8)。2 个成分之间的 KIT 基因型没有差异。然而,2 例伊马替尼耐药肿瘤同时存在 KIT 外显子 11 和外显子 13 突变。荧光原位杂交显示,在 CD117 阴性成分中,与 CD117 阳性区域相比,有 3 例存在 1 个 KIT 基因缺失,2 例存在 KIT 基因低水平扩增。在 5 例检测的病例中,有 1 例存在 TP53 突变,存在于 2 个成分中。总之,GIST 的去分化可以是初发的,也可以是在慢性伊马替尼暴露后发生的,可能是诊断陷阱。这种现象与额外的 KIT 突变无关,但可能是由于遗传不稳定性引起的,表现为杂合性丢失或 KIT 低水平扩增。