Korean Bioinformation Center (KOBIC), Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea.
PLoS One. 2013;8(2):e55596. doi: 10.1371/journal.pone.0055596. Epub 2013 Feb 6.
Deep sequencing techniques provide a remarkable opportunity for comprehensive understanding of tumorigenesis at the molecular level. As omics studies become popular, integrative approaches need to be developed to move from a simple cataloguing of mutations and changes in gene expression to dissecting the molecular nature of carcinogenesis at the systemic level and understanding the complex networks that lead to cancer development.
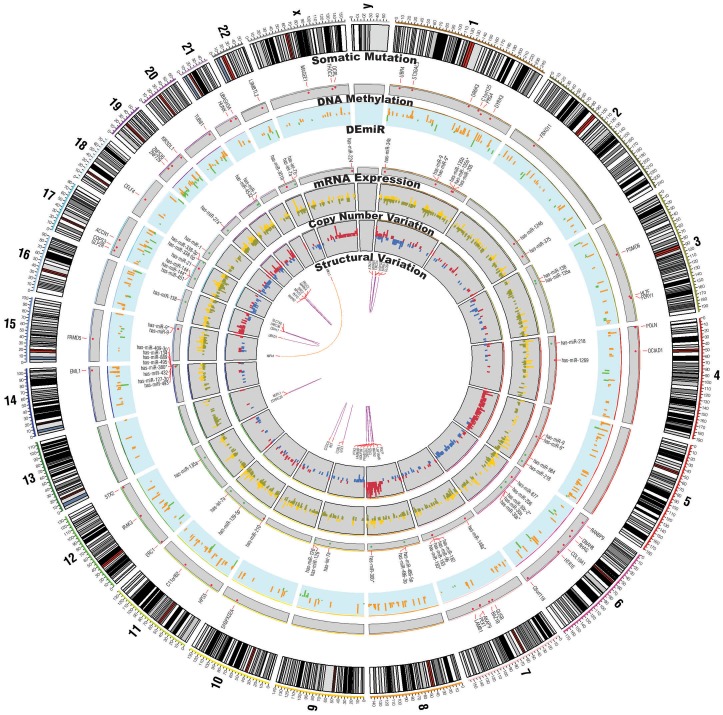
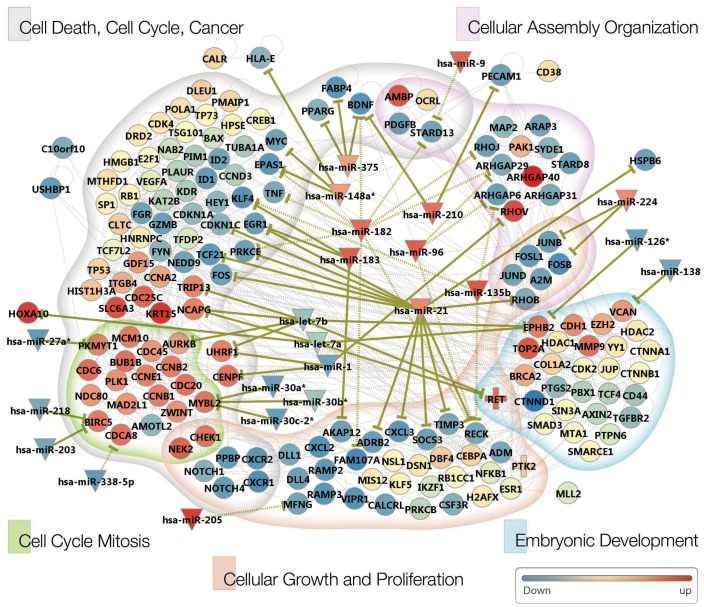
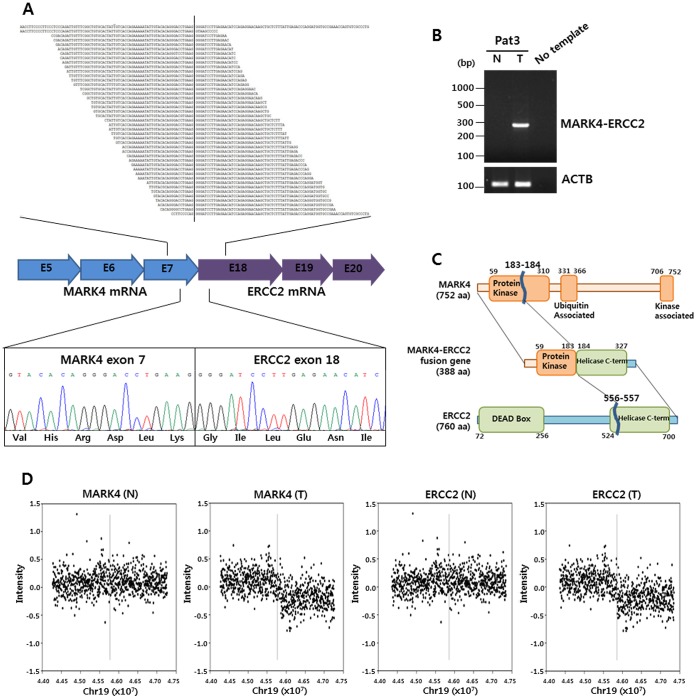
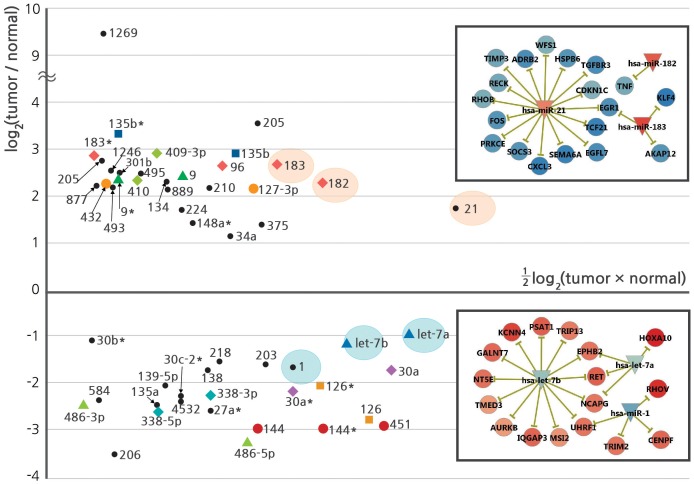
Here, we describe a high-throughput, multi-dimensional sequencing study of primary lung adenocarcinoma tumors and adjacent normal tissues of six Korean female never-smoker patients. Our data encompass results from exome-seq, RNA-seq, small RNA-seq, and MeDIP-seq. We identified and validated novel genetic aberrations, including 47 somatic mutations and 19 fusion transcripts. One of the fusions involves the c-RET gene, which was recently reported to form fusion genes that may function as drivers of carcinogenesis in lung cancer patients. We also characterized gene expression profiles, which we integrated with genomic aberrations and gene regulations into functional networks. The most prominent gene network module that emerged indicates that disturbances in G2/M transition and mitotic progression are causally linked to tumorigenesis in these patients. Also, results from the analysis strongly suggest that several novel microRNA-target interactions represent key regulatory elements of the gene network.
Our study not only provides an overview of the alterations occurring in lung adenocarcinoma at multiple levels from genome to transcriptome and epigenome, but also offers a model for integrative genomics analysis and proposes potential target pathways for the control of lung adenocarcinoma.
深度测序技术为在分子水平上全面了解肿瘤发生提供了极好的机会。随着组学研究的普及,需要开发整合方法,从简单地对突变和基因表达变化进行编目,发展到在系统水平上剖析致癌的分子性质,并理解导致癌症发展的复杂网络。
在这里,我们描述了对 6 名韩国女性从不吸烟的原发性肺腺癌肿瘤和相邻正常组织进行的高通量、多维测序研究。我们的数据包括外显子组测序、RNA-seq、小 RNA-seq 和 MeDIP-seq 的结果。我们鉴定并验证了新的遗传异常,包括 47 个体细胞突变和 19 个融合转录本。其中一个融合涉及 c-RET 基因,最近有报道称该基因融合形成的融合基因可能在肺癌患者的致癌作用中起驱动作用。我们还对基因表达谱进行了特征描述,并将其与基因组异常和基因调控整合到功能网络中。出现的最突出的基因网络模块表明,G2/M 转换和有丝分裂进程中的干扰与这些患者的肿瘤发生有因果关系。此外,分析结果强烈表明,几个新的 microRNA-靶相互作用代表了基因网络的关键调节元件。
我们的研究不仅提供了从基因组到转录组和表观基因组的多个水平上肺腺癌发生变化的概述,还提供了一个整合基因组学分析的模型,并提出了控制肺腺癌的潜在靶向途径。