Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA.
Cell. 2013 Feb 28;152(5):984-96. doi: 10.1016/j.cell.2013.01.038.
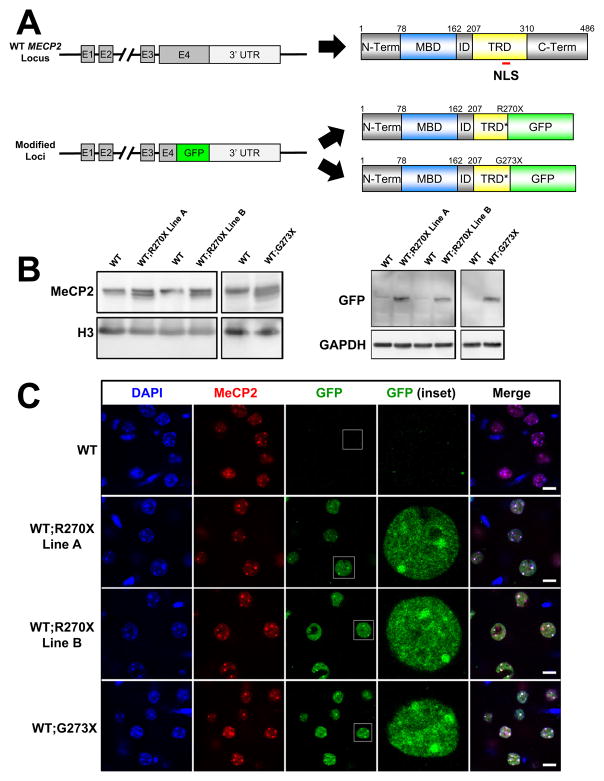
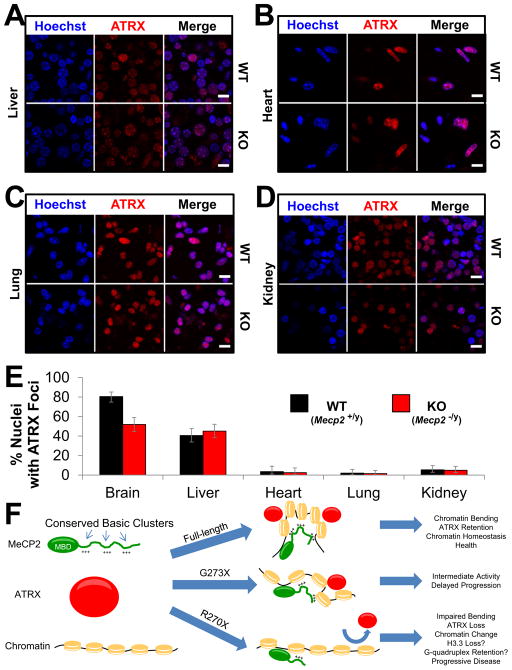
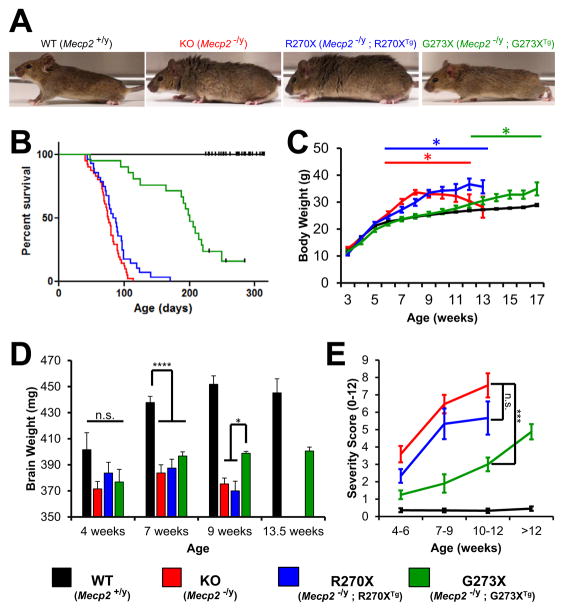
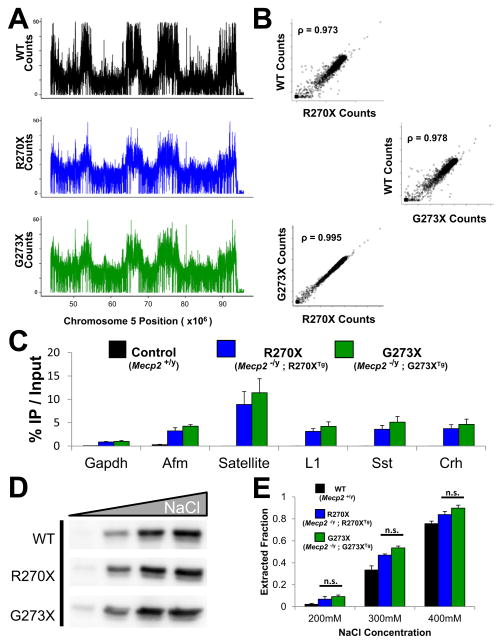
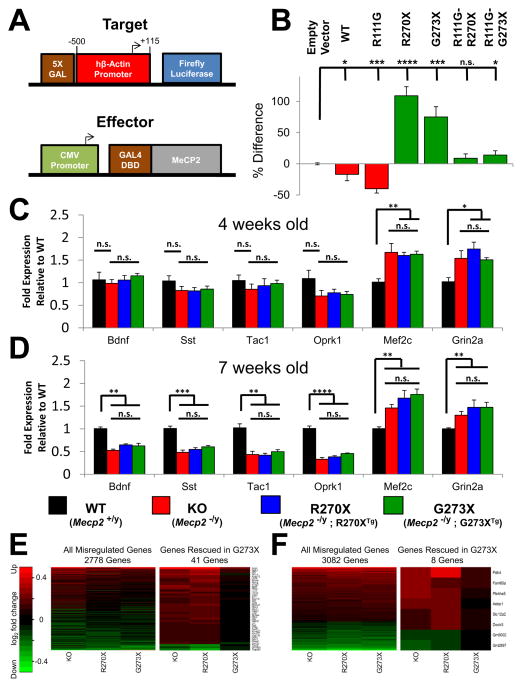
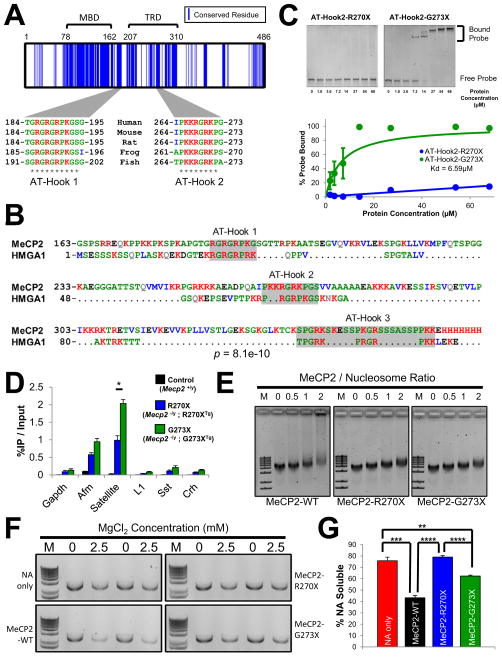
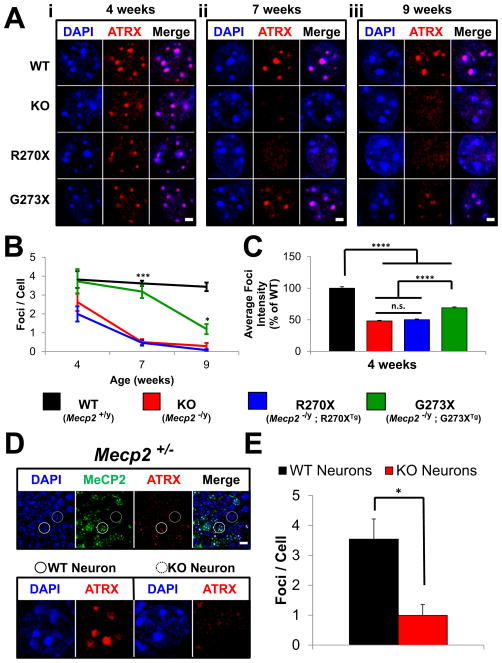
Mutations in the X-linked MECP2 cause Rett syndrome, a devastating neurological disorder typified by a period of apparently normal development followed by loss of cognitive and psychomotor skills. Data from rare male patients suggest symptom onset and severity can be influenced by the location of the mutation, with amino acids 270 and 273 marking the difference between neonatal encephalopathy and death, on the one hand, and survival with deficits on the other. We therefore generated two mouse models expressing either MeCP2-R270X or MeCP2-G273X. The mice developed phenotypes at strikingly different rates and showed differential ATRX nuclear localization within the nervous system, over time, coinciding with phenotypic progression. We discovered that MeCP2 contains three AT-hook-like domains over a stretch of 250 amino acids, like HMGA DNA-bending proteins; one conserved AT-hook is disrupted in MeCP2-R270X, lending further support to the notion that one of MeCP2's key functions is to alter chromatin structure.
X 连锁的 MECP2 基因突变导致雷特综合征,这是一种严重的神经发育障碍,其特征是在明显正常发育之后出现认知和精神运动技能丧失。来自罕见男性患者的数据表明,突变的位置可以影响症状的发作和严重程度,一方面,氨基酸 270 和 273 标志着新生儿脑病和死亡与生存缺陷之间的区别;另一方面。因此,我们生成了两种表达 MeCP2-R270X 或 MeCP2-G273X 的小鼠模型。这些小鼠以惊人的不同速度发展出表型,并显示出随着时间的推移,神经系统内 ATRX 的核定位存在差异,这与表型进展一致。我们发现,MeCP2 在 250 个氨基酸的长片段上包含三个 AT 钩样结构域,类似于 HMGA 弯曲 DNA 的蛋白质;MeCP2-R270X 中的一个保守 AT 钩被破坏,这进一步支持了 MeCP2 的一个关键功能是改变染色质结构的观点。