Guo Yan, Samuels David C, Li Jiang, Clark Travis, Li Chung-I, Shyr Yu
Vanderbilt Ingram Cancer Center, Nashville, TN, USA.
ScientificWorldJournal. 2013;2013:895496. doi: 10.1155/2013/895496. Epub 2013 Feb 7.
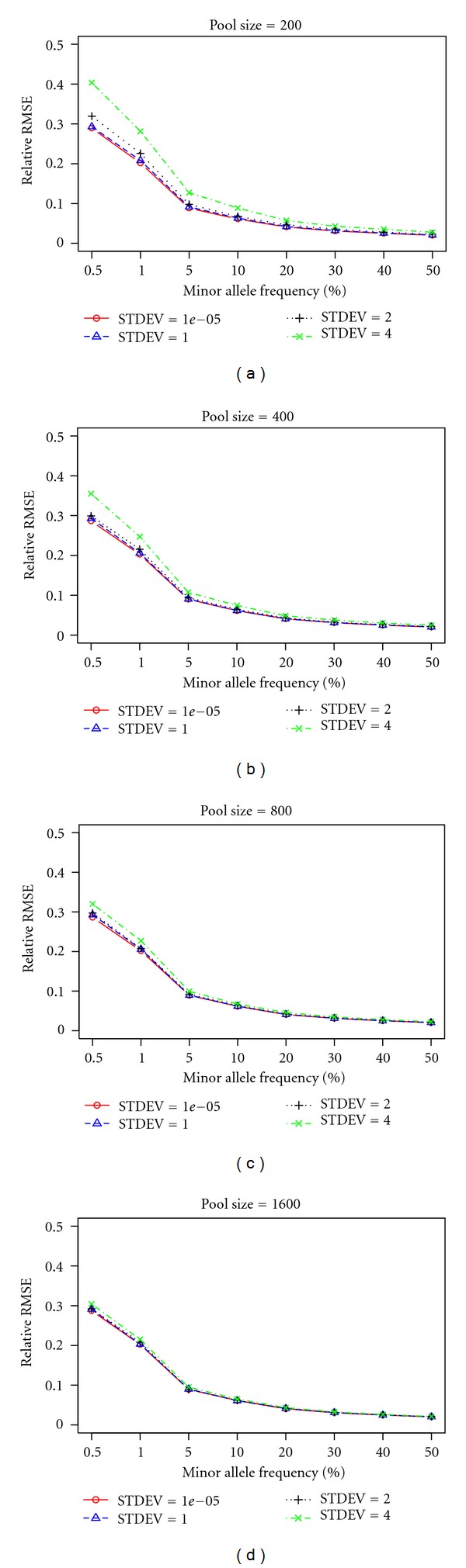
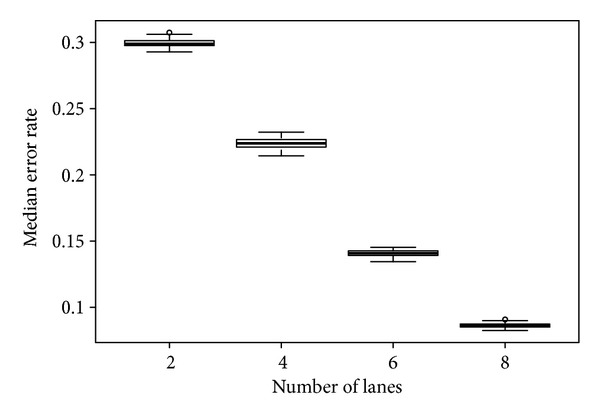
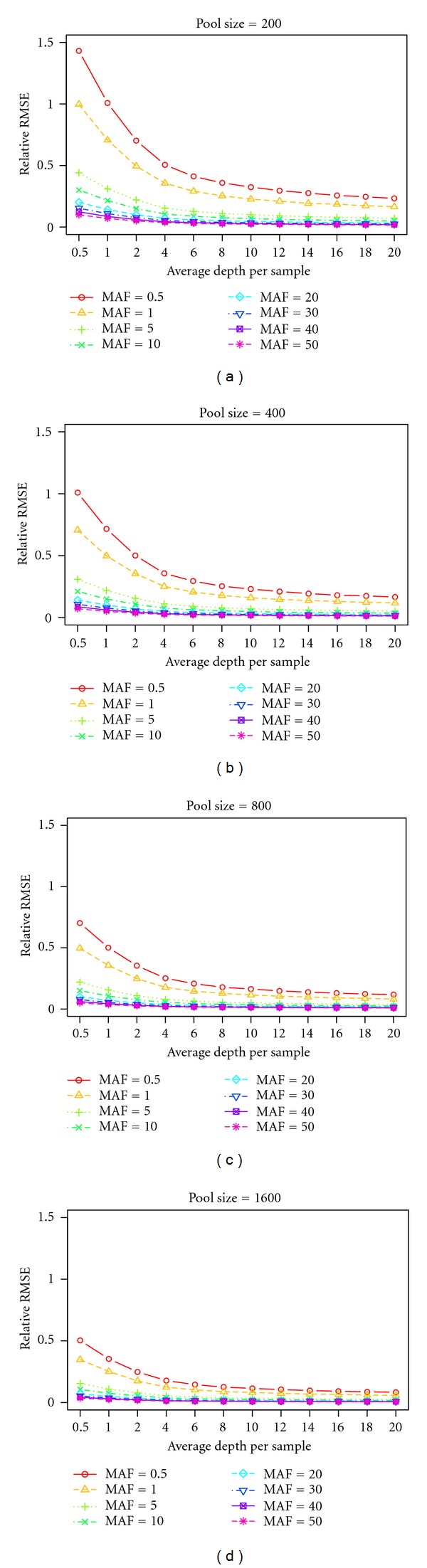
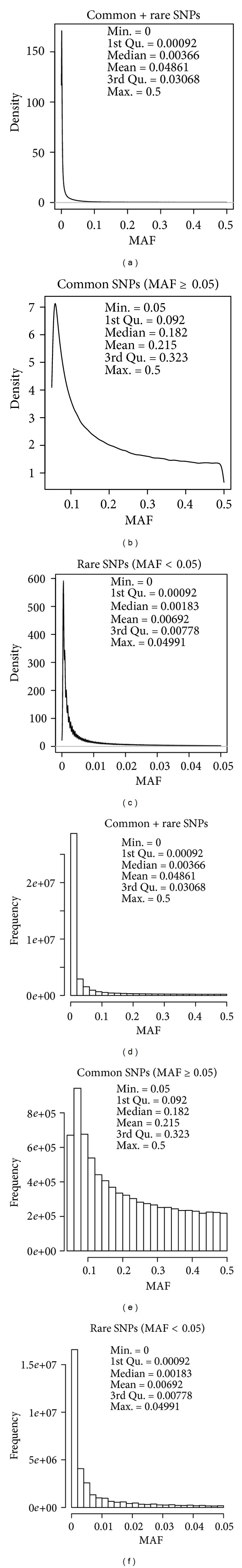
Next-generation sequencing (NGS) technology has provided researchers with opportunities to study the genome in unprecedented detail. In particular, NGS is applied to disease association studies. Unlike genotyping chips, NGS is not limited to a fixed set of SNPs. Prices for NGS are now comparable to the SNP chip, although for large studies the cost can be substantial. Pooling techniques are often used to reduce the overall cost of large-scale studies. In this study, we designed a rigorous simulation model to test the practicability of estimating allele frequency from pooled sequencing data. We took crucial factors into consideration, including pool size, overall depth, average depth per sample, pooling variation, and sampling variation. We used real data to demonstrate and measure reference allele preference in DNAseq data and implemented this bias in our simulation model. We found that pooled sequencing data can introduce high levels of relative error rate (defined as error rate divided by targeted allele frequency) and that the error rate is more severe for low minor allele frequency SNPs than for high minor allele frequency SNPs. In order to overcome the error introduced by pooling, we recommend a large pool size and high average depth per sample.
下一代测序(NGS)技术为研究人员提供了前所未有的详细研究基因组的机会。特别是,NGS被应用于疾病关联研究。与基因分型芯片不同,NGS不限于一组固定的单核苷酸多态性(SNP)。目前NGS的价格与SNP芯片相当,尽管对于大型研究来说成本可能很高。合并技术通常用于降低大规模研究的总体成本。在本研究中,我们设计了一个严格的模拟模型来测试从合并测序数据估计等位基因频率的实用性。我们考虑了关键因素,包括池大小、总体深度、每个样本的平均深度、合并变异和抽样变异。我们使用真实数据来证明和测量DNA测序数据中的参考等位基因偏好,并在我们的模拟模型中实现这种偏差。我们发现,合并测序数据会引入高水平的相对错误率(定义为错误率除以目标等位基因频率),并且对于低次要等位基因频率的SNP,错误率比高次要等位基因频率的SNP更严重。为了克服合并引入的错误,我们建议采用大的池大小和每个样本高的平均深度。