Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China.
BMC Genomics. 2013 Mar 27;14:206. doi: 10.1186/1471-2164-14-206.
Adenosine-to-inosine (A-to-I) RNA editing is recognized as a cellular mechanism for generating both RNA and protein diversity. Inosine base pairs with cytidine during reverse transcription and therefore appears as guanosine during sequencing of cDNA. Current approaches of RNA editing identification largely depend on the comparison between transcriptomes and genomic DNA (gDNA) sequencing datasets from the same individuals, and it has been challenging to identify editing candidates from transcriptomes in the absence of gDNA information.
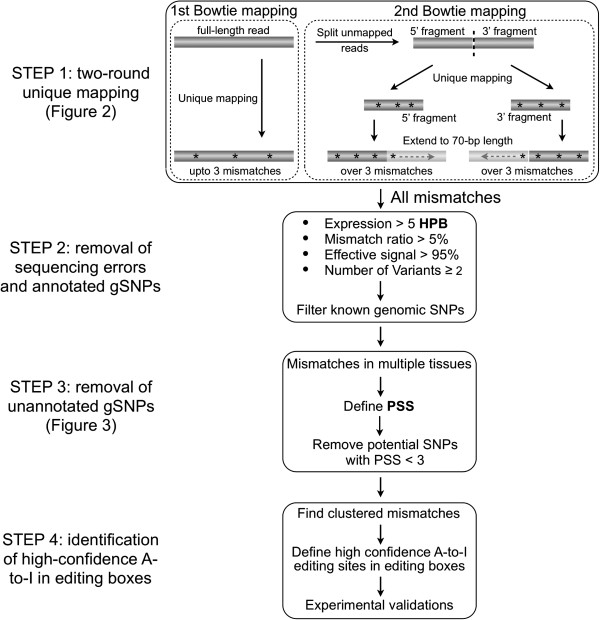
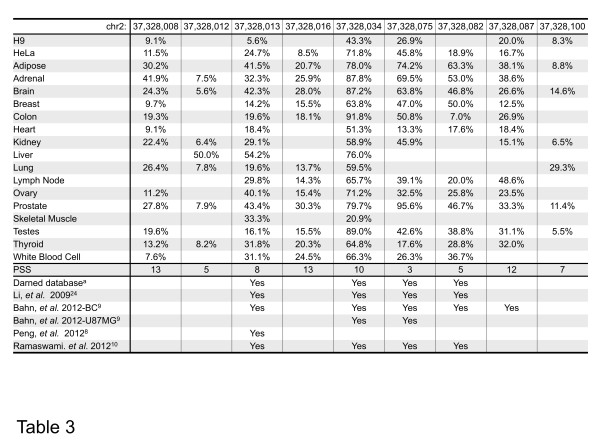
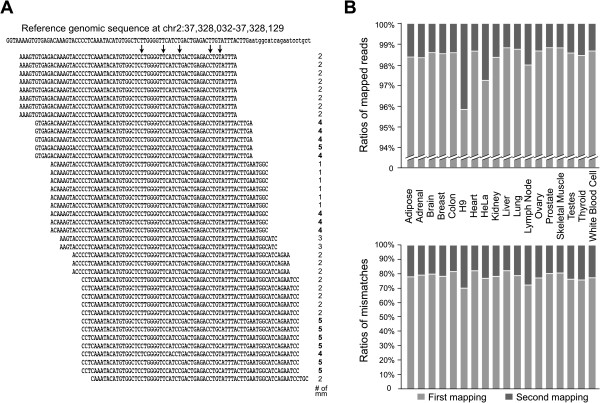
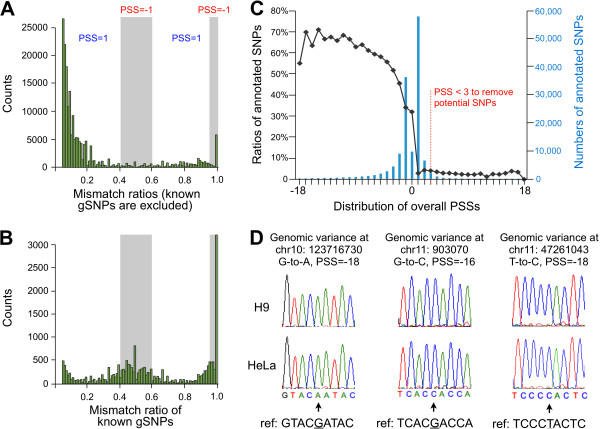
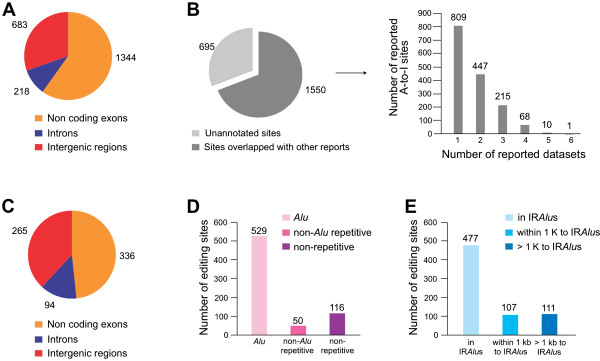
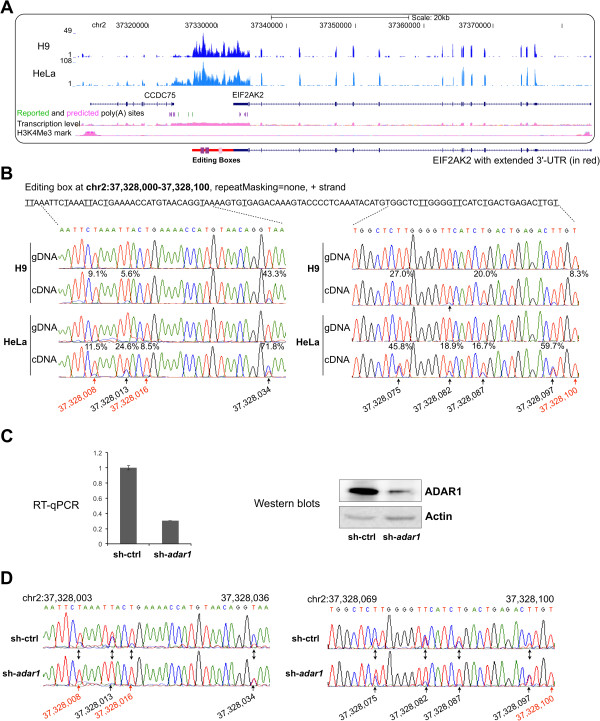
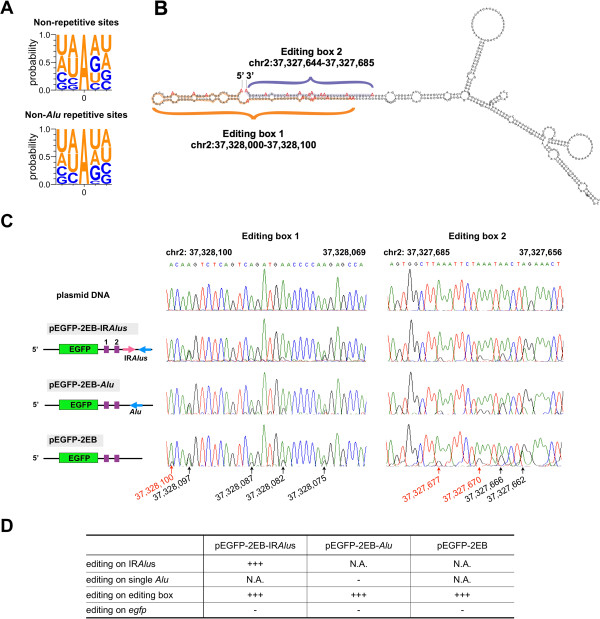
We have developed a new strategy to accurately predict constitutive RNA editing sites from publicly available human RNA-seq datasets in the absence of relevant genomic sequences. Our approach establishes new parameters to increase the ability to map mismatches and to minimize sequencing/mapping errors and unreported genome variations. We identified 695 novel constitutive A-to-I editing sites that appear in clusters (named "editing boxes") in multiple samples and which exhibit spatial and dynamic regulation across human tissues. Some of these editing boxes are enriched in non-repetitive regions lacking inverted repeat structures and contain an extremely high conversion frequency of As to Is. We validated a number of editing boxes in multiple human cell lines and confirmed that ADAR1 is responsible for the observed promiscuous editing events in non-repetitive regions, further expanding our knowledge of the catalytic substrate of A-to-I RNA editing by ADAR enzymes.
The approach we present here provides a novel way of identifying A-to-I RNA editing events by analyzing only RNA-seq datasets. This method has allowed us to gain new insights into RNA editing and should also aid in the identification of more constitutive A-to-I editing sites from additional transcriptomes.
腺嘌呤到次黄嘌呤(A-to-I)的 RNA 编辑被认为是一种产生 RNA 和蛋白质多样性的细胞机制。在反转录过程中,次黄嘌呤与胞嘧啶配对,因此在 cDNA 的测序中表现为鸟嘌呤。目前的 RNA 编辑识别方法主要依赖于对同一个体的转录组和基因组 DNA(gDNA)测序数据集之间的比较,并且在没有 gDNA 信息的情况下,从转录组中识别编辑候选物一直具有挑战性。
我们开发了一种新策略,可在没有相关基因组序列的情况下,从公开的人类 RNA-seq 数据集准确预测组成性 RNA 编辑位点。我们的方法建立了新的参数,以提高映射错配的能力,并最大限度地减少测序/映射错误和未报告的基因组变异。我们鉴定了 695 个新的组成性 A-to-I 编辑位点,这些位点在多个样本中出现在簇中(命名为“编辑框”),并在人类组织中表现出空间和动态调节。其中一些编辑框富集在没有反向重复结构的非重复区域,并包含极高的 As 到 Is 转换频率。我们在多个人类细胞系中验证了多个编辑框,并证实 ADAR1 负责非重复区域中观察到的混杂编辑事件,进一步扩展了我们对 ADAR 酶介导的 A-to-I RNA 编辑催化底物的认识。
我们在这里提出的方法通过仅分析 RNA-seq 数据集提供了一种识别 A-to-I RNA 编辑事件的新方法。这种方法使我们能够深入了解 RNA 编辑,并应该有助于从其他转录组中识别更多的组成性 A-to-I 编辑位点。