Department of Medicine, Center for Immunology, University of Minnesota, Minneapolis, Minnesota, USA.
Diabetes. 2013 Aug;62(8):2859-69. doi: 10.2337/db12-1475. Epub 2013 Apr 4.
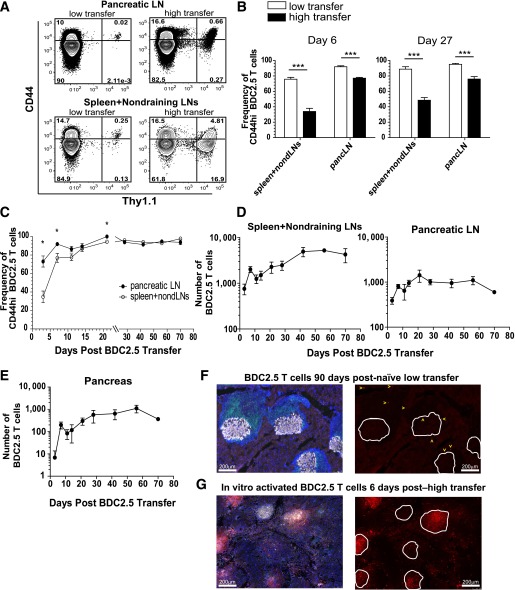
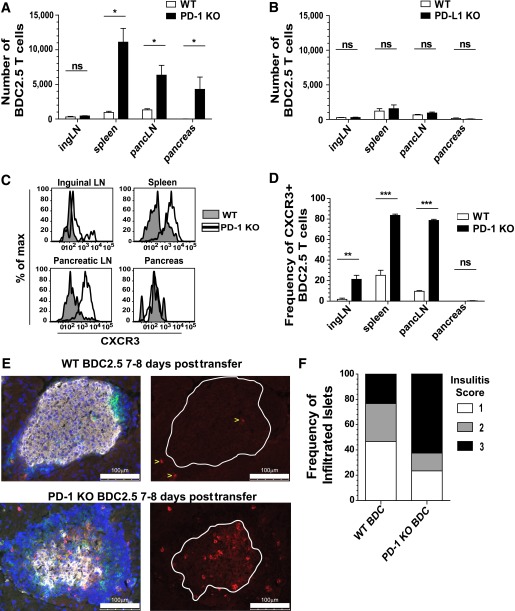
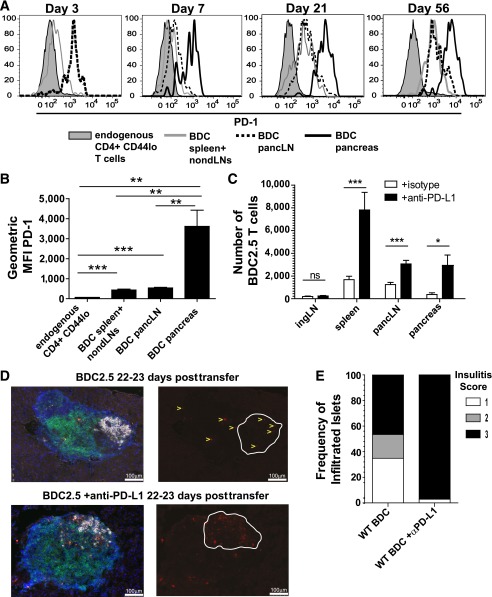
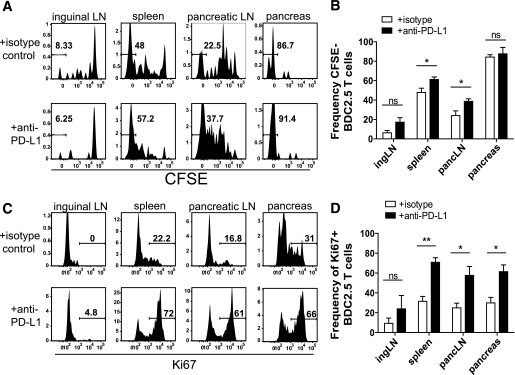
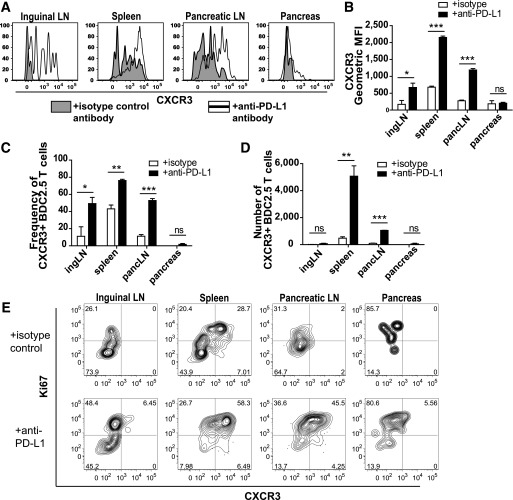
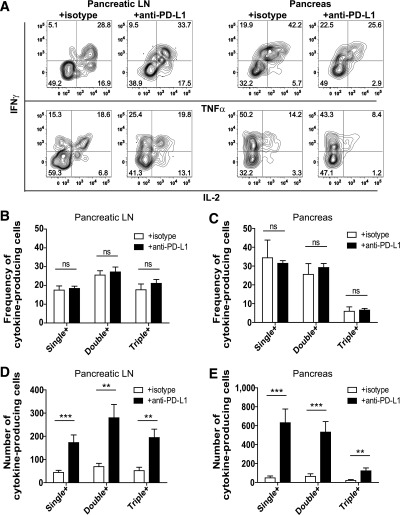
The inhibitory receptor programmed death-1 (PD-1) constrains type 1 diabetes (T1D) in the nonobese diabetic (NOD) mouse. However, how PD-1 influences diabetogenic CD4(+) T cells during natural diabetes is not fully understood. To address this question, we developed a novel model to investigate antigen-specific CD4(+) T cells under physiological conditions in vivo. We transferred a low number of naïve CD4(+) T cells from the BDC2.5 mouse into prediabetic NOD mice to mimic a physiological precursor frequency and allowed the cells to become primed by endogenous autoantigen. Transferred BDC2.5 T cells became activated, differentiated into T-bet(+) IFN-γ-producing cells, and infiltrated the pancreas. In this model, loss of PD-1, but not programmed death ligand-1 (PD-L1), on the antigen-specific CD4(+) T cell resulted in increased cell numbers in the spleen, pancreas-draining lymph node, and pancreas. PD-1 deficiency also increased expression of the chemokine receptor CXCR3. Lastly, histological data showed that a loss of PD-1 caused BDC2.5 cells to penetrate deep into the islet core, resulting in conversion from peri-insulitis to destructive insulitis. These data support a model by which PD-1 regulates islet-reactive CD4(+) T cells in a cell intrinsic manner by suppressing proliferation, inhibiting infiltration of the pancreas, and limiting diabetes.
抑制受体程序性死亡-1(PD-1)限制了非肥胖型糖尿病(NOD)小鼠的 1 型糖尿病(T1D)。然而,PD-1 在自然发生糖尿病的情况下如何影响致糖尿病性 CD4(+) T 细胞尚未完全了解。为了解决这个问题,我们开发了一种新的模型来研究体内生理条件下的抗原特异性 CD4(+) T 细胞。我们将来自 BDC2.5 小鼠的少量幼稚 CD4(+) T 细胞转移到糖尿病前期的 NOD 小鼠中,以模拟生理前体频率,并允许细胞被内源性自身抗原激活。转移的 BDC2.5 T 细胞被激活,分化为 T-bet(+) IFN-γ产生细胞,并浸润胰腺。在这种模型中,抗原特异性 CD4(+) T 细胞上 PD-1 的缺失,但不是程序性死亡配体-1(PD-L1)的缺失,导致脾脏、胰腺引流淋巴结和胰腺中的细胞数量增加。PD-1 缺乏还增加了趋化因子受体 CXCR3 的表达。最后,组织学数据表明,PD-1 的缺失导致 BDC2.5 细胞穿透胰岛核心,从胰岛周围炎转变为破坏性胰岛炎。这些数据支持了一种模型,即 PD-1 通过抑制增殖、抑制胰腺浸润和限制糖尿病,以细胞内方式调节胰岛反应性 CD4(+) T 细胞。