Department of Craniofacial Biology, School of Dental Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA.
Oncogenesis. 2013 Mar 25;2(3):e39. doi: 10.1038/oncsis.2013.4.
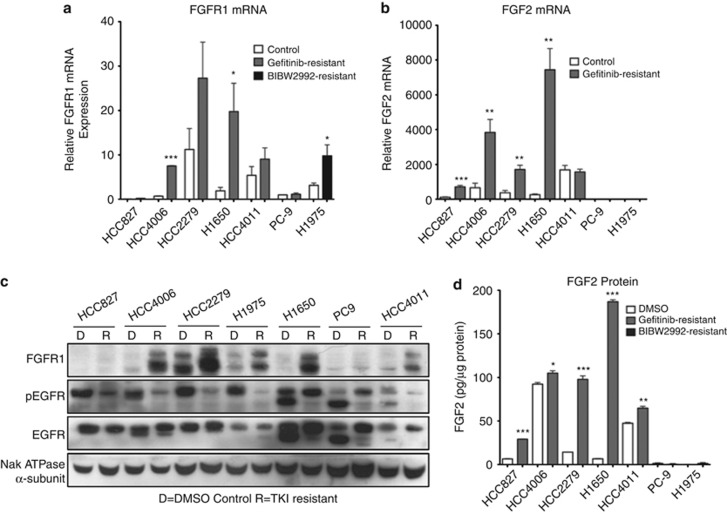
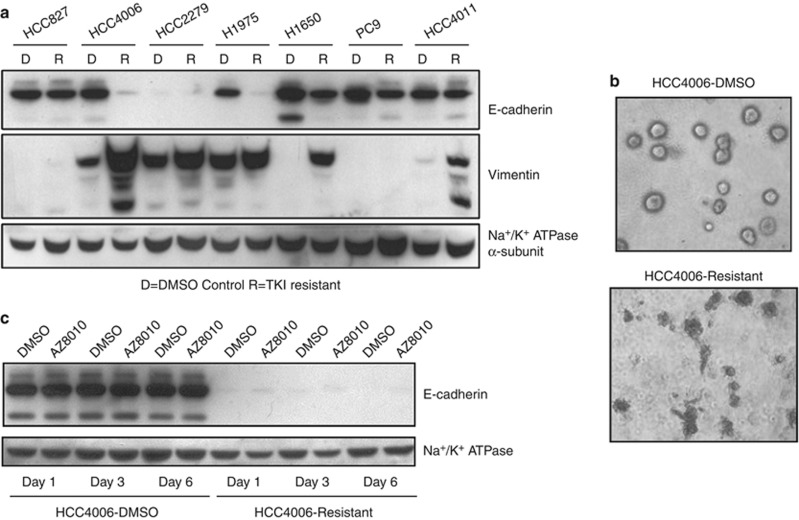
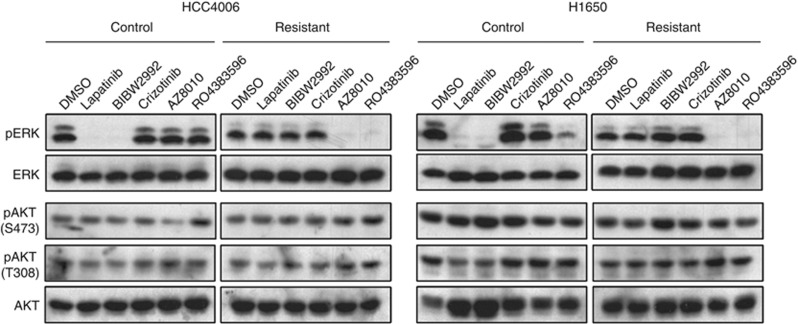
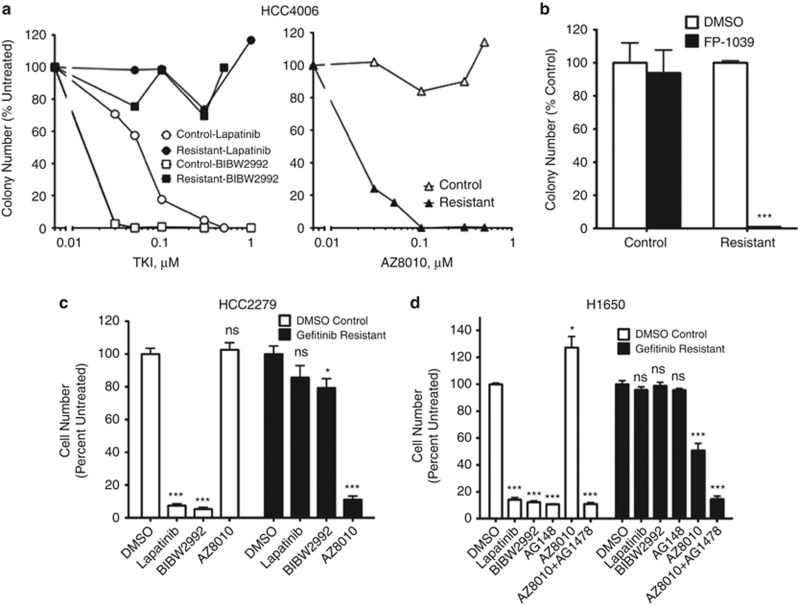
Despite initial and often dramatic responses of epidermal growth factor receptor (EGFR)-addicted lung tumors to the EGFR-specific tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib, nearly all develop resistance and relapse. To explore novel mechanisms mediating acquired resistance, we employed non-small-cell lung cancer (NSCLC) cell lines bearing activating mutations in EGFR and rendered them resistant to EGFR-specific TKIs through chronic adaptation in tissue culture. In addition to previously observed resistance mechanisms including EGFR-T790M 'gate-keeper' mutations and MET amplification, a subset of the seven chronically adapted NSCLC cell lines including HCC4006, HCC2279 and H1650 cells exhibited marked induction of fibroblast growth factor (FGF) 2 and FGF receptor 1 (FGFR1) mRNA and protein. Also, adaptation to EGFR-specific TKIs was accompanied by an epithelial to mesenchymal transition (EMT) as assessed by changes in CDH1, VIM, ZEB1 and ZEB2 expression and altered growth properties in Matrigel. In adapted cell lines exhibiting increased FGF2 and FGFR1 expression, measures of growth and signaling, but not EMT, were blocked by FGFR-specific TKIs, an FGF-ligand trap and FGFR1 silencing with RNAi. In parental HCC4006 cells, cell growth was strongly inhibited by gefitinib, although drug-resistant clones progress within 10 days. Combined treatment with gefitinib and AZD4547, an FGFR-specific TKI, prevented the outgrowth of drug-resistant clones. Thus, induction of FGF2 and FGFR1 following chronic adaptation to EGFR-specific TKIs provides a novel autocrine receptor tyrosine kinase-driven bypass pathway in a subset of lung cancer cell lines that are initially sensitive to EGFR-specific TKIs. The findings support FGFR-specific TKIs as potentially valuable additions to existing targeted therapeutic strategies with EGFR-specific TKIs to prevent or delay acquired resistance in EGFR-driven NSCLC.
尽管表皮生长因子受体 (EGFR) 依赖性肺肿瘤对 EGFR 特异性酪氨酸激酶抑制剂 (TKI),如吉非替尼和厄洛替尼,最初有明显的反应,甚至经常有戏剧性的反应,但几乎所有肿瘤最终都会产生耐药性并复发。为了探索介导获得性耐药的新机制,我们使用了携带 EGFR 激活突变的非小细胞肺癌 (NSCLC) 细胞系,并通过在组织培养中进行慢性适应使这些细胞系对 EGFR 特异性 TKI 产生耐药性。除了先前观察到的耐药机制,包括 EGFR-T790M“门控”突变和 MET 扩增,在七种慢性适应的 NSCLC 细胞系中,包括 HCC4006、HCC2279 和 H1650 细胞,亚组细胞显著诱导成纤维细胞生长因子 (FGF) 2 和 FGF 受体 1 (FGFR1) mRNA 和蛋白的表达。此外,正如 CDH1、VIM、ZEB1 和 ZEB2 表达的变化以及 Matrigel 中生长特性的改变所评估的那样,对 EGFR 特异性 TKI 的适应伴随着上皮间质转化 (EMT)。在表达 FGF2 和 FGFR1 增加的适应细胞系中,生长和信号的测定,但不是 EMT,被 FGFR 特异性 TKI、FGF 配体陷阱和 RNAi 沉默 FGFR1 阻断。在亲本 HCC4006 细胞中,吉非替尼强烈抑制细胞生长,尽管耐药克隆在 10 天内就会发展。吉非替尼与 FGFR 特异性 TKI AZD4547 的联合治疗可防止耐药克隆的生长。因此,在对 EGFR 特异性 TKI 进行慢性适应后,FGF2 和 FGFR1 的诱导为一组最初对 EGFR 特异性 TKI 敏感的肺癌细胞系提供了一种新的自分泌受体酪氨酸激酶驱动的旁路途径。这些发现支持 FGFR 特异性 TKI 作为现有针对 EGFR 驱动 NSCLC 的 EGFR 特异性 TKI 靶向治疗策略的潜在有价值的补充,以预防或延迟获得性耐药。