Department of Molecular Genetics and Microbiology, College of Medicine, University of Florida, Gainesville, FL, USA.
BMC Bioinformatics. 2013 Apr 4;14:116. doi: 10.1186/1471-2105-14-116.
Next generation transcriptome sequencing (RNA-Seq) is emerging as a powerful experimental tool for the study of alternative splicing and its regulation, but requires ad-hoc analysis methods and tools. PASTA (Patterned Alignments for Splicing and Transcriptome Analysis) is a splice junction detection algorithm specifically designed for RNA-Seq data, relying on a highly accurate alignment strategy and on a combination of heuristic and statistical methods to identify exon-intron junctions with high accuracy.
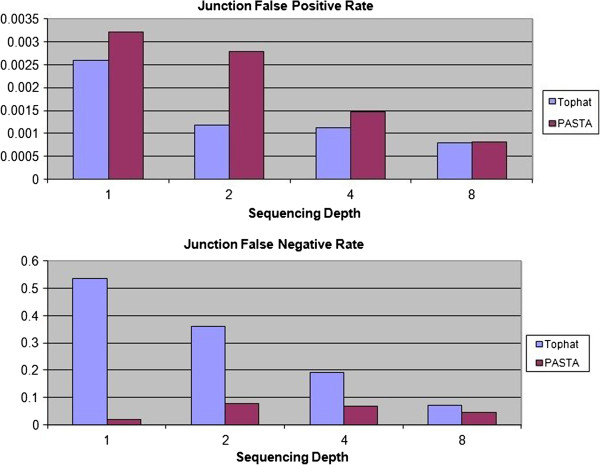
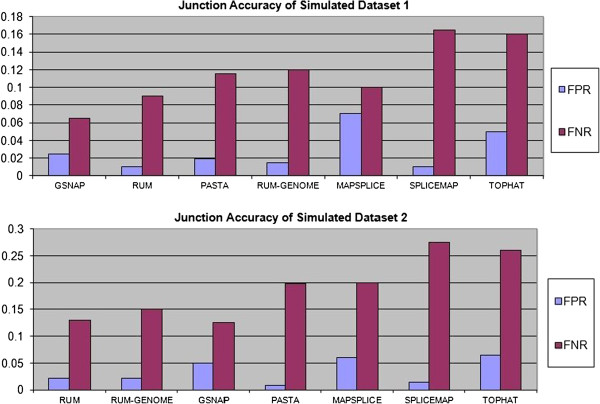
Comparisons against TopHat and other splice junction prediction software on real and simulated datasets show that PASTA exhibits high specificity and sensitivity, especially at lower coverage levels. Moreover, PASTA is highly configurable and flexible, and can therefore be applied in a wide range of analysis scenarios: it is able to handle both single-end and paired-end reads, it does not rely on the presence of canonical splicing signals, and it uses organism-specific regression models to accurately identify junctions.
PASTA is a highly efficient and sensitive tool to identify splicing junctions from RNA-Seq data. Compared to similar programs, it has the ability to identify a higher number of real splicing junctions, and provides highly annotated output files containing detailed information about their location and characteristics. Accurate junction data in turn facilitates the reconstruction of the splicing isoforms and the analysis of their expression levels, which will be performed by the remaining modules of the PASTA pipeline, still under development. Use of PASTA can therefore enable the large-scale investigation of transcription and alternative splicing.
下一代转录组测序(RNA-Seq)作为一种研究可变剪接及其调控的强大实验工具正在兴起,但需要特定的分析方法和工具。PASTA(用于剪接和转录组分析的模式对齐)是一种专门为 RNA-Seq 数据设计的剪接接头检测算法,它依赖于高度准确的对齐策略和启发式和统计方法的组合,以高精度识别外显子-内含子接头。
与 TopHat 和其他剪接接头预测软件在真实和模拟数据集上的比较表明,PASTA 表现出高特异性和灵敏度,特别是在较低的覆盖水平下。此外,PASTA具有高度可配置性和灵活性,因此可以应用于广泛的分析场景:它能够处理单端和双端读取,不依赖于规范剪接信号的存在,并且使用特定于生物体的回归模型来准确识别接头。
PASTA 是一种从 RNA-Seq 数据中识别剪接接头的高效、敏感工具。与类似的程序相比,它能够识别更多真实的剪接接头,并提供包含其位置和特征详细信息的高度注释输出文件。准确的接头数据反过来又促进了剪接异构体的重建和它们的表达水平的分析,这将由 PASTA 管道的其余模块(仍在开发中)来执行。因此,PASTA 的使用可以实现转录和可变剪接的大规模研究。