Institute of Cell Biology and Neurobiology, Charité University Medicine, Berlin, Germany.
Orphanet J Rare Dis. 2013 Apr 15;8:59. doi: 10.1186/1750-1172-8-59.
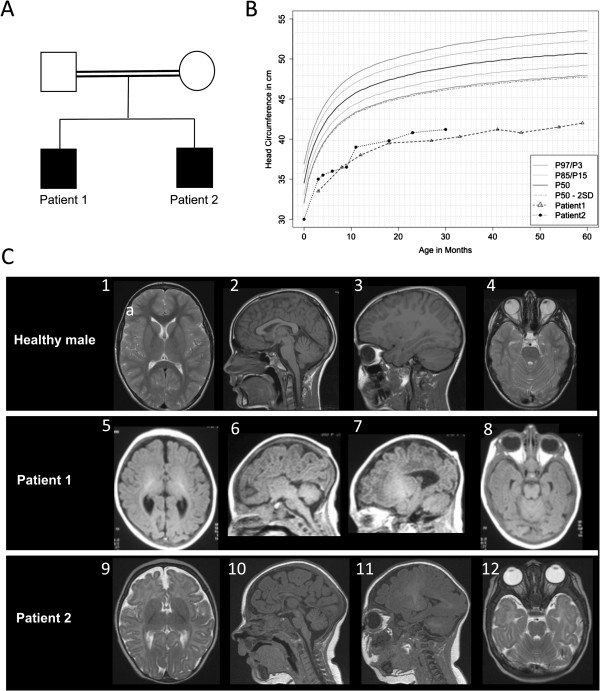
Primary autosomal recessive microcephaly (MCPH) is a rare neurodevelopmental disorder that results in severe microcephaly at birth with pronounced reduction in brain volume, particularly of the neocortex, simplified cortical gyration and intellectual disability. Homozygous mutations in the Cyclin-dependent kinase 5 regulatory subunit-associated protein 2 gene CDK5RAP2 are the cause of MCPH3. Despite considerable interest in MCPH as a model disorder for brain development, the underlying pathomechanism has not been definitively established and only four pedigrees with three CDK5RAP2 mutations have been reported. Specifically for MCPH3, no detailed radiological or histological descriptions exist.
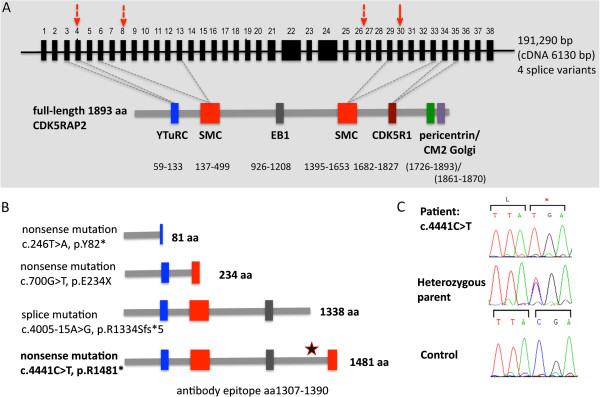
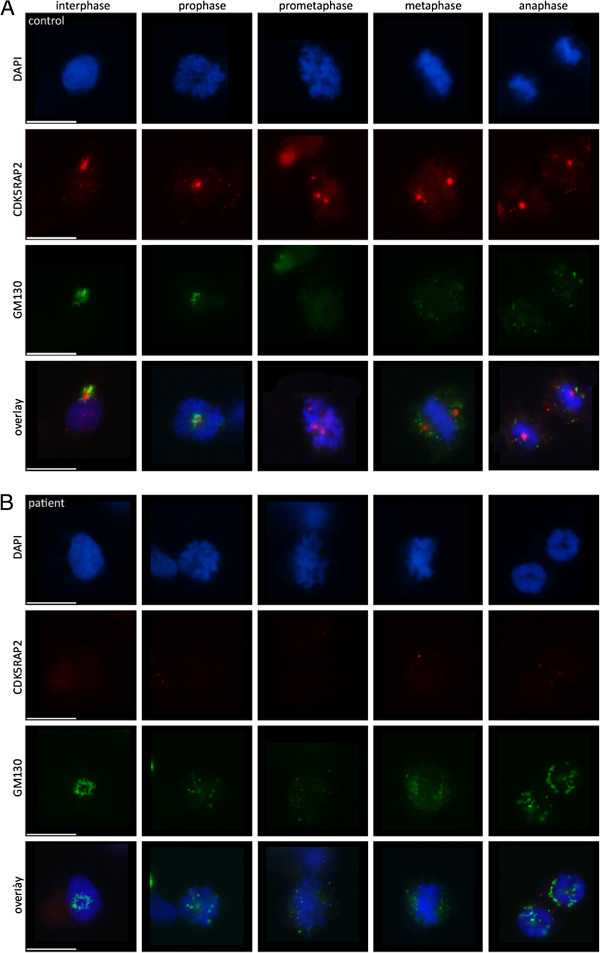
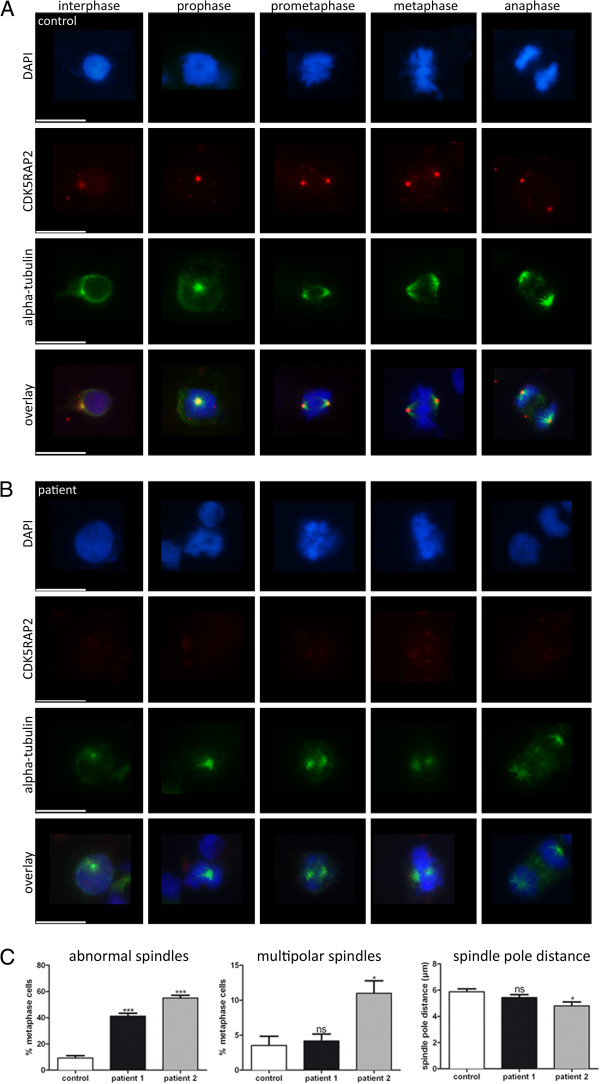
METHODS/RESULTS: We sought to characterize the clinical and radiological features and pathological cellular processes that contribute to the human MCPH3 phenotype. Haplotype analysis using microsatellite markers around the MCPH1-7 and PNKP loci in an Italian family with two sons with primary microcephaly, revealed possible linkage to the MCPH3 locus. Sequencing of the coding exons and exon/intron splice junctions of the CDK5RAP2 gene identified homozygosity for the novel nonsense mutation, c.4441C > T (p.Arg1481*), in both affected sons. cMRI showed microcephaly, simplified gyral pattern and hypogenesis of the corpus callosum. The cellular phenotype was assessed in EBV-transformed lymphocyte cell lines established from the two affected sons and compared with healthy male controls. CDK5RAP2 protein levels were below detection level in immortalized lymphocytes from the patients. Moreover, mitotic spindle defects and disrupted γ-tubulin localization to the centrosome were apparent.
These results suggest that spindle defects and a disruption of centrosome integrity play an important role in the development of microcephaly in MCPH3.
常染色体隐性原发性小头畸形(MCPH)是一种罕见的神经发育障碍,导致出生时严重的小头畸形,伴有明显的脑容量减少,特别是新皮质,皮质回旋简化和智力残疾。周期蛋白依赖性激酶 5 调节亚单位相关蛋白 2 基因(CDK5RAP2)的纯合突变是 MCPH3 的病因。尽管人们对 MCPH 作为脑发育障碍模型非常感兴趣,但潜在的病理机制尚未明确确定,并且仅报道了四个具有三个 CDK5RAP2 突变的家系。具体来说,对于 MCPH3,没有详细的影像学或组织学描述。
方法/结果:我们试图描述导致人类 MCPH3 表型的临床和影像学特征以及病理细胞过程。在一个意大利家庭中,使用 MCPH1-7 和 PNKP 基因座周围的微卫星标记进行单体型分析,该家庭中有两个患有原发性小头畸形的儿子,显示出与 MCPH3 基因座可能的连锁。对 CDK5RAP2 基因的编码外显子和外显子/内含子剪接接头进行测序,发现两个受影响的儿子均为纯合性新型无意义突变,c.4441C>T(p.Arg1481*)。cMRI 显示小头畸形,脑回模式简化和胼胝体发育不良。评估了从两个受影响的儿子中建立的 EBV 转化的淋巴细胞系中的细胞表型,并与健康男性对照进行了比较。患者的永生化淋巴细胞中 CDK5RAP2 蛋白水平低于检测水平。此外,明显存在有丝分裂纺锤体缺陷和 γ-微管蛋白向中心体的定位中断。
这些结果表明,纺锤体缺陷和中心体完整性的破坏在 MCPH3 中微小头畸形的发育中起重要作用。