Will Sebastian, Siebauer Michael F, Heyne Steffen, Engelhardt Jan, Stadler Peter F, Backofen Rolf
Bioinformatics Group, Department of Computer Science, and Interdisciplinary Center for Bioinformatics, University of Leipzig, Härtelstraße 16 -18, Leipzig D-04107, Germany.
Bioinformatics Group, Department of Computer Science, Albert-Ludwigs-Universität Freiburg, Georges-Köhler-Allee 106, Freiburg D-79110, Germany.
Algorithms Mol Biol. 2013 Apr 20;8:14. doi: 10.1186/1748-7188-8-14. eCollection 2013.
The search for distant homologs has become an import issue in genome annotation. A particular difficulty is posed by divergent homologs that have lost recognizable sequence similarity. This same problem also arises in the recognition of novel members of large classes of RNAs such as snoRNAs or microRNAs that consist of families unrelated by common descent. Current homology search tools for structured RNAs are either based entirely on sequence similarity (such as blast or hmmer) or combine sequence and secondary structure. The most prominent example of the latter class of tools is Infernal. Alternatives are descriptor-based methods. In most practical applications published to-date, however, the information contained in covariance models or manually prescribed search patterns is dominated by sequence information. Here we ask two related questions: (1) Is secondary structure alone informative for homology search and the detection of novel members of RNA classes? (2) To what extent is the thermodynamic propensity of the target sequence to fold into the correct secondary structure helpful for this task?
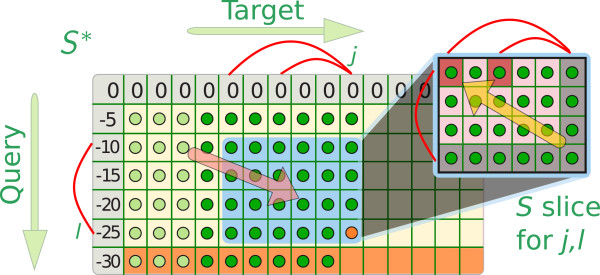
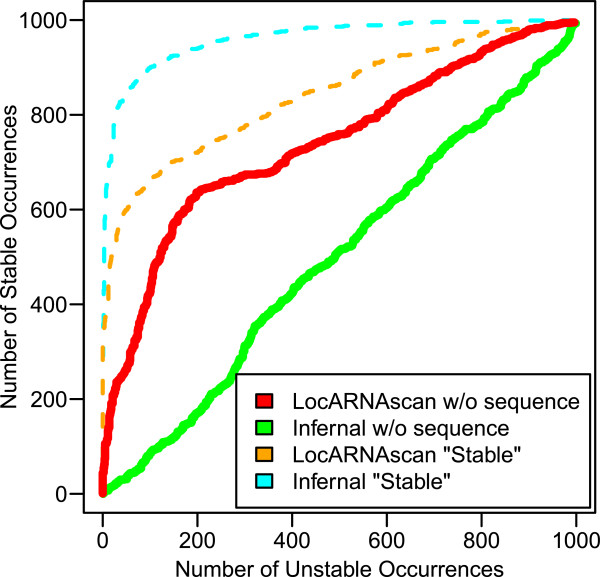
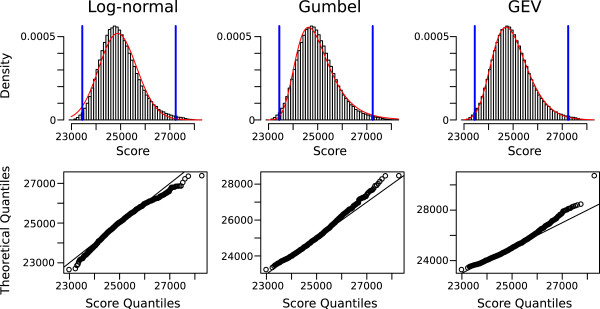
Sequence-structure alignment can be used as an alternative search strategy. In this scenario, the query consists of a base pairing probability matrix, which can be derived either from a single sequence or from a multiple alignment representing a set of known representatives. Sequence information can be optionally added to the query. The target sequence is pre-processed to obtain local base pairing probabilities. As a search engine we devised a semi-global scanning variant of LocARNA's algorithm for sequence-structure alignment. The LocARNAscan tool is optimized for speed and low memory consumption. In benchmarking experiments on artificial data we observe that the inclusion of thermodynamic stability is helpful, albeit only in a regime of extremely low sequence information in the query. We observe, furthermore, that the sensitivity is bounded in particular by the limited accuracy of the predicted local structures of the target sequence.
Although we demonstrate that a purely structure-based homology search is feasible in principle, it is unlikely to outperform tools such as Infernal in most application scenarios, where a substantial amount of sequence information is typically available. The LocARNAscan approach will profit, however, from high throughput methods to determine RNA secondary structure. In transcriptome-wide applications, such methods will provide accurate structure annotations on the target side.
Source code of the free software LocARNAscan 1.0 and supplementary data are available at http://www.bioinf.uni-leipzig.de/Software/LocARNAscan.
寻找远源同源物已成为基因组注释中的一个重要问题。具有发散性的同源物失去了可识别的序列相似性,这带来了一个特殊的困难。在识别诸如snoRNA或microRNA等由非共同起源的家族组成的大类RNA的新成员时,同样的问题也会出现。当前用于结构化RNA的同源性搜索工具要么完全基于序列相似性(如blast或hmmer),要么结合序列和二级结构。后一类工具中最突出的例子是Infernal。还有基于描述符的方法。然而,在迄今为止发表的大多数实际应用中,协方差模型或手动规定的搜索模式中包含的信息主要由序列信息主导。在这里,我们提出两个相关问题:(1)仅二级结构对于同源性搜索和RNA类新成员的检测是否具有信息性?(2)目标序列折叠成正确二级结构的热力学倾向在多大程度上有助于这项任务?
序列-结构比对可以用作一种替代搜索策略。在这种情况下,查询由一个碱基配对概率矩阵组成,该矩阵可以从单个序列或从代表一组已知代表的多序列比对中推导出来。序列信息可以选择性地添加到查询中。对目标序列进行预处理以获得局部碱基配对概率。作为搜索引擎,我们设计了LocARNA算法的一种半全局扫描变体用于序列-结构比对。LocARNAscan工具针对速度和低内存消耗进行了优化。在人工数据的基准实验中,我们观察到纳入热力学稳定性是有帮助的,尽管仅在查询中序列信息极低的情况下。此外,我们观察到灵敏度尤其受到目标序列预测局部结构有限准确性的限制。
虽然我们证明了纯基于结构的同源性搜索原则上是可行的,但在大多数通常有大量序列信息可用的应用场景中,它不太可能优于Infernal等工具。然而,LocARNAscan方法将受益于高通量方法来确定RNA二级结构。在全转录组应用中,此类方法将在目标方面提供准确的结构注释。
免费软件LocARNAscan 1.0的源代码和补充数据可在http://www.bioinf.uni-leipzig.de/Software/LocARNAscan获取。