Department of Molecular and Cell Biology, Henry M. Goldman School of Dental Medicine, Boston University, MA, USA.
FEBS J. 2013 Jun;280(12):2888-99. doi: 10.1111/febs.12292. Epub 2013 May 16.
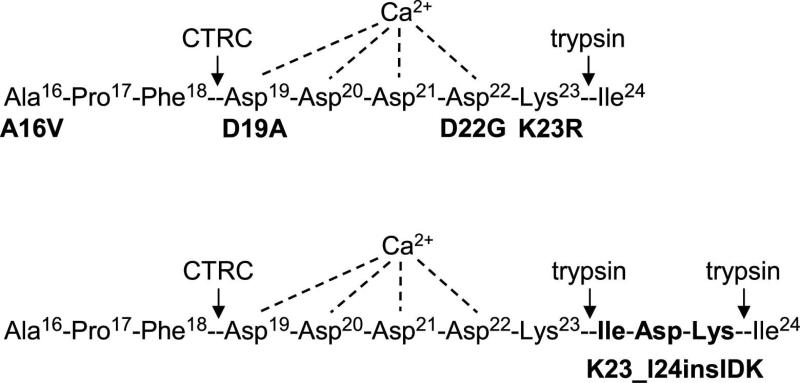
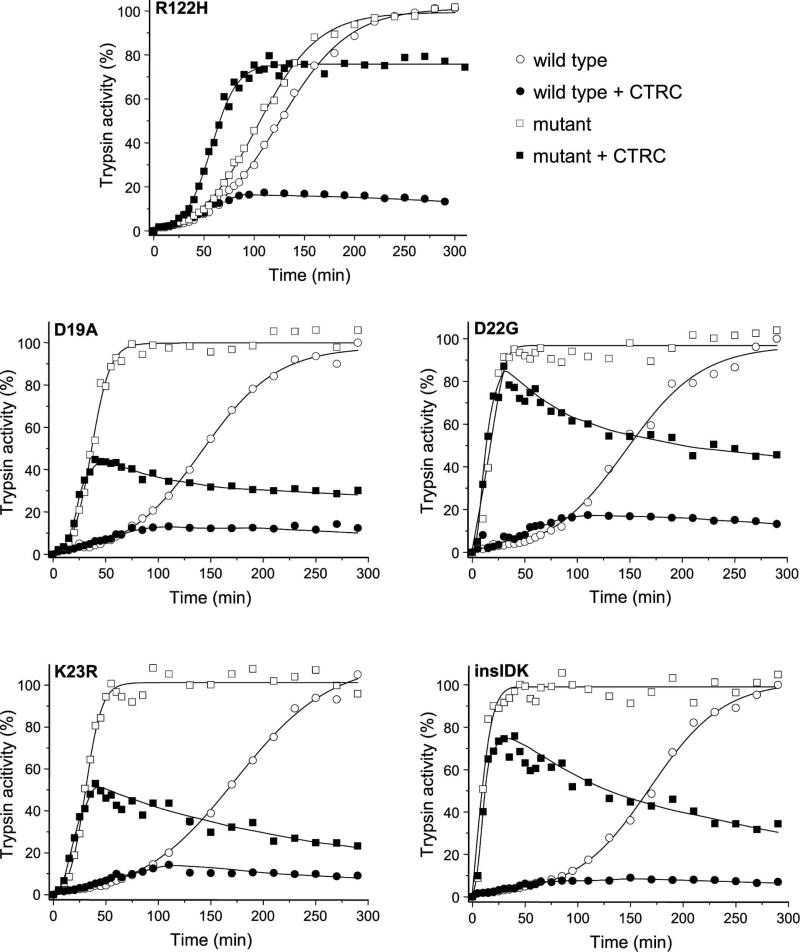
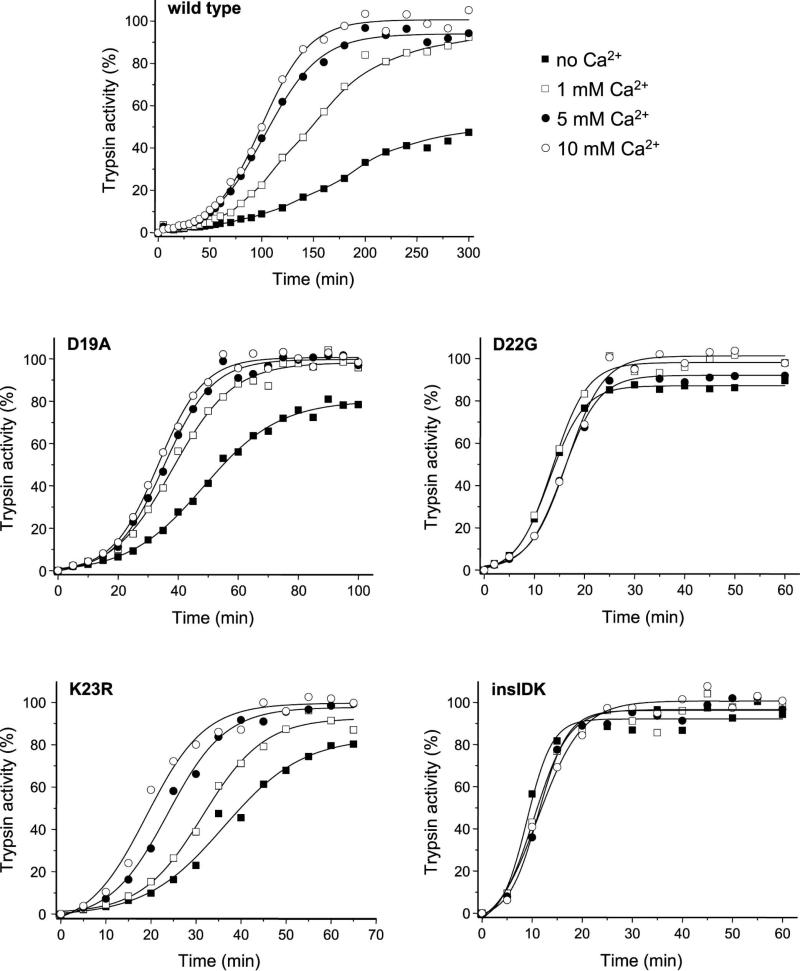
Mutations in human cationic trypsinogen cause hereditary pancreatitis by altering its proteolytic regulation of activation and degradation by chymotrypsin C (CTRC). CTRC stimulates trypsinogen autoactivation by processing the activation peptide to a shorter form, but also promotes degradation by cleaving the calcium-binding loop in trypsinogen. Mutations render trypsinogen resistant to CTRC-mediated degradation and/or increase processing of the activation peptide by CTRC. Here we demonstrate that the activation peptide mutations D19A, D22G, K23R and K23_I24insIDK robustly increased the rate of trypsinogen autoactivation, both in the presence and absence of CTRC. Degradation of the mutants by CTRC was unchanged, and processing of the activation peptide was increased fourfold in the D19A mutant only. Surprisingly, however, this increased processing had only a minimal effect on autoactivation. The tetra-aspartate motif in the trypsinogen activation peptide binds calcium (KD of ~ 1.6 mM), which stimulates autoactivation. Unexpectedly, calcium binding was not compromised by any of the activation peptide mutations. Despite normal binding, autoactivation of mutants D22G and K23_I24insIDK was not stimulated by calcium. Finally, the activation peptide mutants exhibited reduced secretion from transfected cells, and secreted trypsinogen levels were inversely proportional with autoactivation rates. We conclude that D19A, D22G, K23R and K23_I24insIDK form a mechanistically distinct subset of hereditary pancreatitis-associated mutations that exert their effect primarily through direct stimulation of autoactivation, independently of CTRC. The potentially severe clinical impact of the markedly increased autoactivation is offset by diminished secretion, resulting in a clinical phenotype that is indistinguishable from typical hereditary pancreatitis.
人类阳离子胰蛋白酶原中的突变通过改变其对糜蛋白酶 C (CTRC)的蛋白水解调节,导致遗传性胰腺炎。CTRC 通过将激活肽加工成较短的形式来刺激胰蛋白酶原的自动激活,但也通过切割胰蛋白酶原中的钙结合环来促进其降解。突变使胰蛋白酶原对 CTRC 介导的降解具有抗性和/或增加 CTRC 对激活肽的加工。在这里,我们证明激活肽突变 D19A、D22G、K23R 和 K23_I24insIDK 均显著增加了胰蛋白酶原的自动激活速率,无论是在存在还是不存在 CTRC 的情况下。突变体被 CTRC 降解的情况没有改变,只有 D19A 突变体中激活肽的加工增加了四倍。然而,令人惊讶的是,这种增加的加工对自动激活只有很小的影响。胰蛋白酶原激活肽中的四天门冬氨酸基序结合钙(KD 约为 1.6mM),这刺激了自动激活。出乎意料的是,任何激活肽突变都没有损害钙结合。尽管结合正常,突变体 D22G 和 K23_I24insIDK 的自动激活不受钙的刺激。最后,激活肽突变体显示从转染细胞中的分泌减少,并且分泌的胰蛋白酶原水平与自动激活速率成反比。我们得出结论,D19A、D22G、K23R 和 K23_I24insIDK 形成了一组机制上不同的遗传性胰腺炎相关突变,它们主要通过直接刺激自动激活来发挥作用,而不依赖于 CTRC。自动激活明显增加所带来的潜在严重临床影响被分泌减少所抵消,导致与典型遗传性胰腺炎无法区分的临床表型。