Molecular Biology Graduate Program, Graduate School, University of Colorado Anschuts Medical Campus, Aurora, CO, USA.
Department of Craniofacial Biology, School of Dental Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA.
Oncogene. 2014 Mar 27;33(13):1670-9. doi: 10.1038/onc.2013.115. Epub 2013 Apr 22.
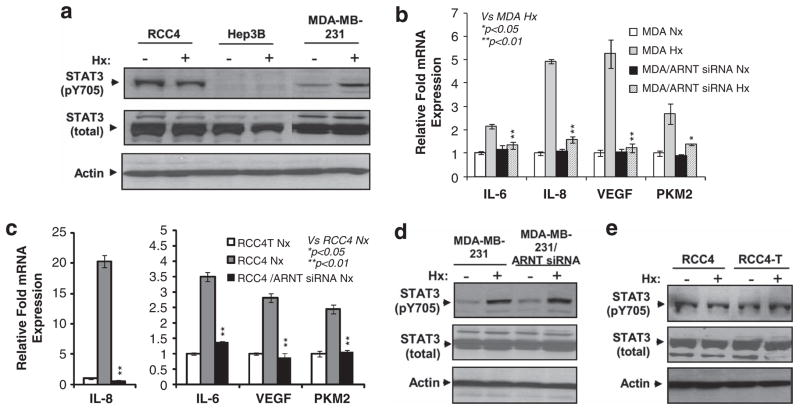
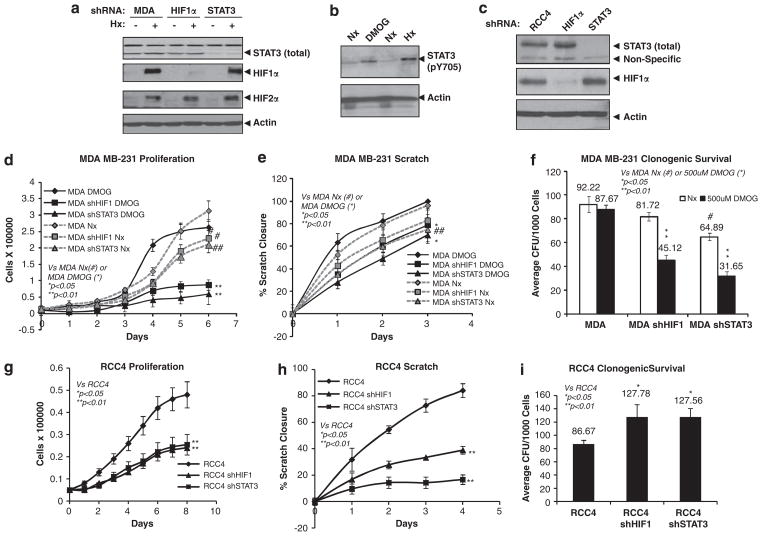
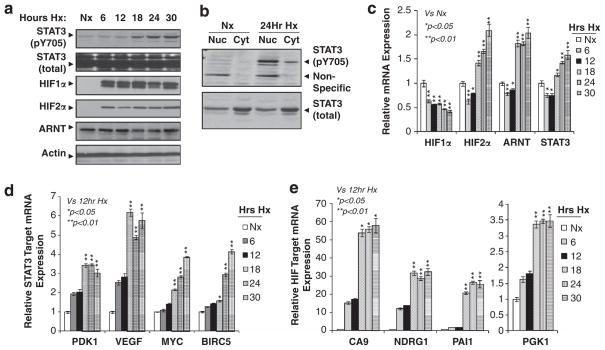
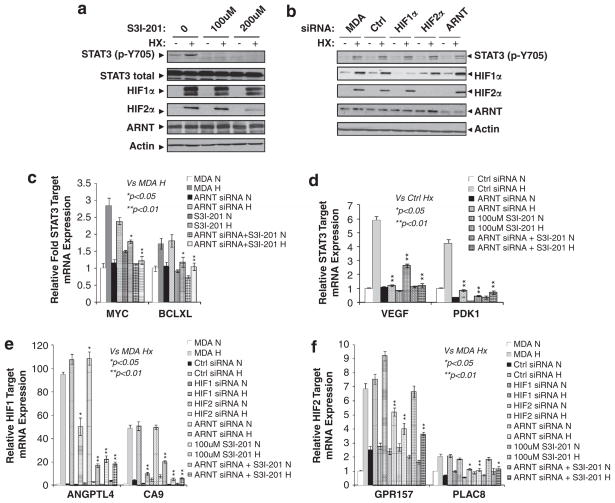
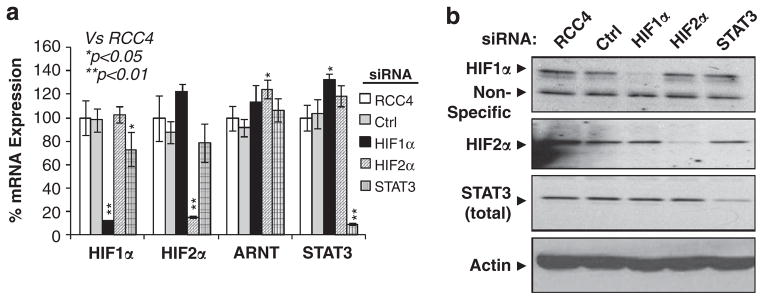
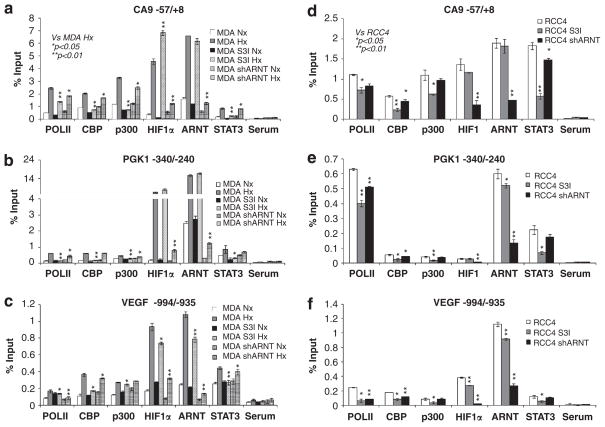
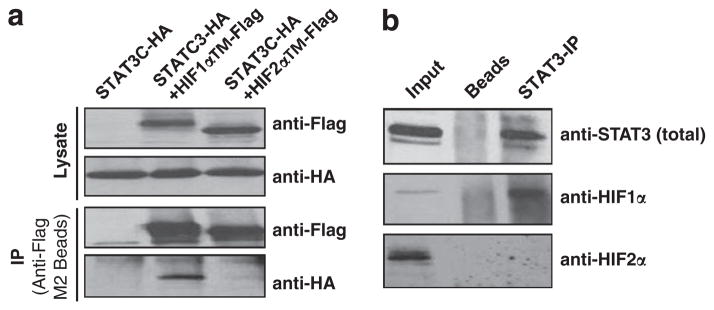
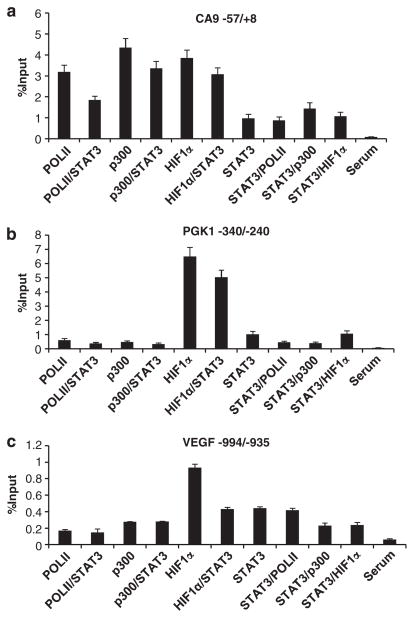
Solid tumors often exhibit simultaneously inflammatory and hypoxic microenvironments. The 'signal transducer and activator of transcription-3' (STAT3)-mediated inflammatory response and the hypoxia-inducible factor (HIF)-mediated hypoxia response have been independently shown to promote tumorigenesis through the activation of HIF or STAT3 target genes and to be indicative of a poor prognosis in a variety of tumors. We report here for the first time that STAT3 is involved in the HIF1, but not HIF2-mediated hypoxic transcriptional response. We show that inhibiting STAT3 activity in MDA-MB-231 and RCC4 cells by a STAT3 inhibitor or STAT3 small interfering RNA significantly reduces the levels of HIF1, but not HIF2 target genes in spite of normal levels of hypoxia-inducible transcription factor 1α (HIF1α) and HIF2α protein. Mechanistically, STAT3 activates HIF1 target genes by binding to HIF1 target gene promoters, interacting with HIF1α protein and recruiting coactivators CREB binding protein (CBP) and p300, and RNA polymerase II (Pol II) to form enhanceosome complexes that contain HIF1α, STAT3, CBP, p300 and RNA Pol II on HIF1 target gene promoters. Functionally, the effect of STAT3 knockdown on proliferation, motility and clonogenic survival of tumor cells in vitro is phenocopied by HIF1α knockdown in hypoxic cells, whereas STAT3 knockdown in normoxic cells also reduces cell proliferation, motility and clonogenic survival. This indicates that STAT3 works with HIF1 to activate HIF1 target genes and to drive HIF1-depedent tumorigenesis under hypoxic conditions, but also has HIF-independent activity in normoxic and hypoxic cells. Identifying the role of STAT3 in the hypoxia response provides further data supporting the effectiveness of STAT3 inhibitors in solid tumor treatment owing to their usefulness in inhibiting both the STAT3 and HIF1 pro-tumorigenic signaling pathways in some cancer types.
实体瘤常表现出同时存在炎症和缺氧的微环境。已经独立地表明“信号转导和转录激活因子 3”(STAT3)介导的炎症反应和缺氧诱导因子(HIF)介导的缺氧反应通过激活 HIF 或 STAT3 靶基因促进肿瘤发生,并表明在各种肿瘤中预后不良。我们在这里首次报道 STAT3 参与 HIF1,但不参与 HIF2 介导的缺氧转录反应。我们表明,通过 STAT3 抑制剂或 STAT3 小干扰 RNA 抑制 MDA-MB-231 和 RCC4 细胞中的 STAT3 活性,尽管缺氧诱导转录因子 1α(HIF1α)和 HIF2α 蛋白水平正常,但显著降低 HIF1,但不降低 HIF2 靶基因的水平。在机制上,STAT3 通过与 HIF1 靶基因启动子结合、与 HIF1α 蛋白相互作用并募集共激活因子 CREB 结合蛋白(CBP)和 p300 以及 RNA 聚合酶 II(Pol II)来激活 HIF1 靶基因,形成包含 HIF1α、STAT3、CBP、p300 和 RNA Pol II 的增强子复合物在 HIF1 靶基因启动子上。功能上,在缺氧细胞中 HIF1α 敲低可模拟 STAT3 敲低对肿瘤细胞体外增殖、迁移和集落形成存活的影响,而在常氧细胞中 STAT3 敲低也降低细胞增殖、迁移和集落形成存活。这表明 STAT3 与 HIF1 一起激活 HIF1 靶基因,并在缺氧条件下驱动 HIF1 依赖性肿瘤发生,但在常氧和缺氧细胞中也具有 HIF 非依赖性活性。鉴定 STAT3 在缺氧反应中的作用为 STAT3 抑制剂在实体瘤治疗中的有效性提供了进一步的数据支持,因为它们在一些癌症类型中对抑制 STAT3 和 HIF1 促肿瘤信号通路都有用。