Departamento de Ciencias Biológicas, Facultad de Ciencias Biológicas & Facultad de Medicina, Universidad Andres Bello, Santiago, Chile.
J Cell Mol Med. 2013 Jun;17(6):800-14. doi: 10.1111/jcmm.12066. Epub 2013 May 2.
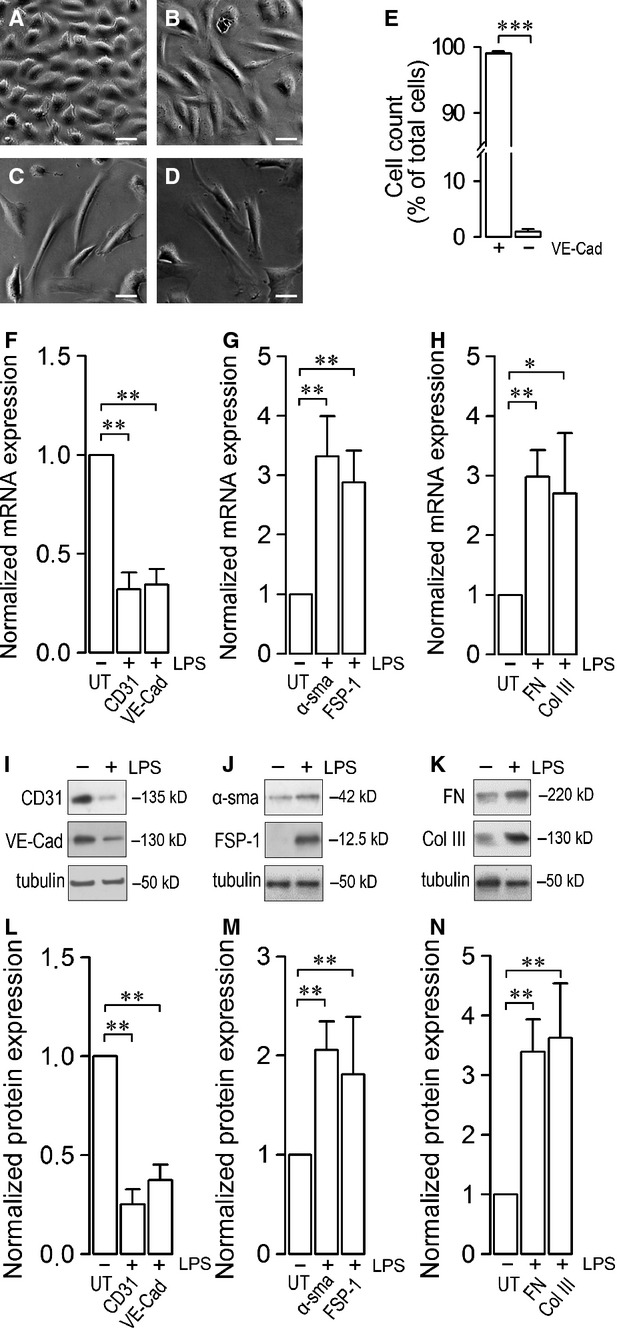
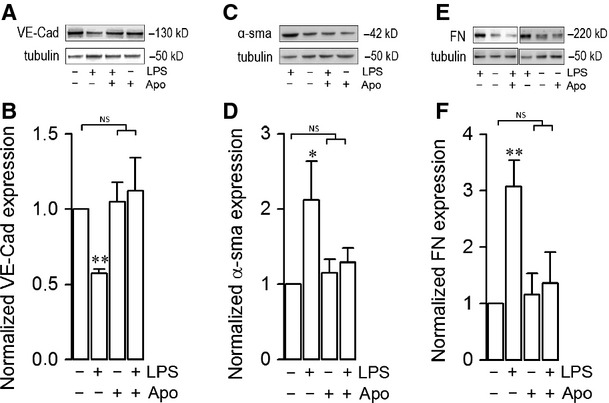
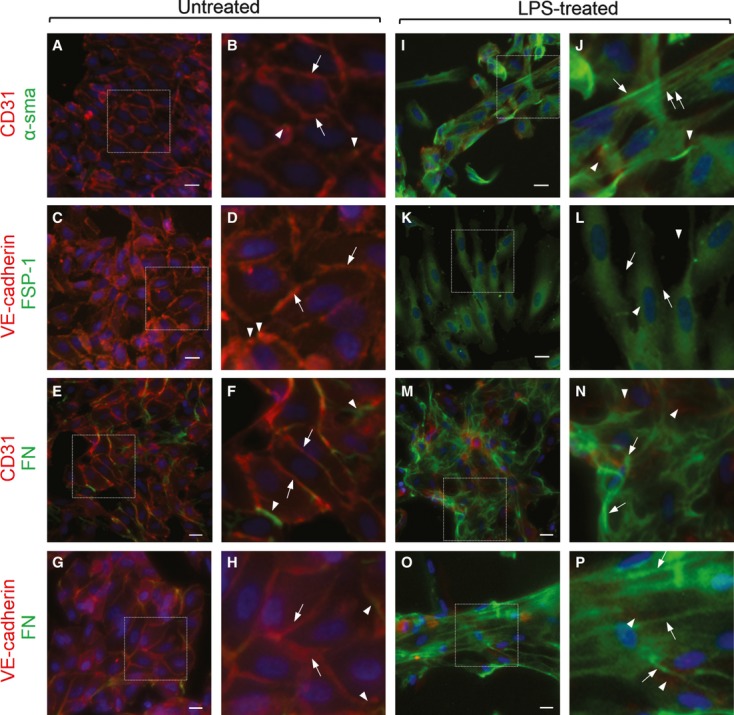
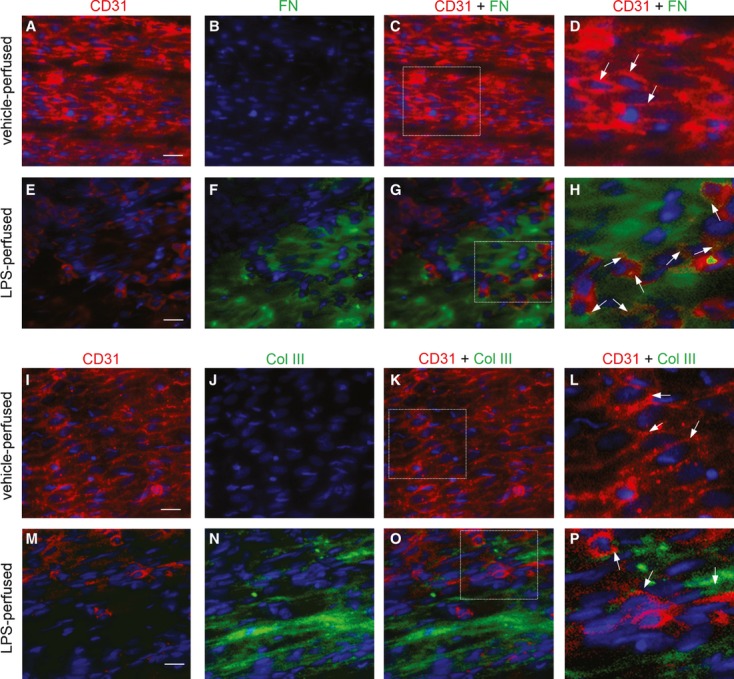
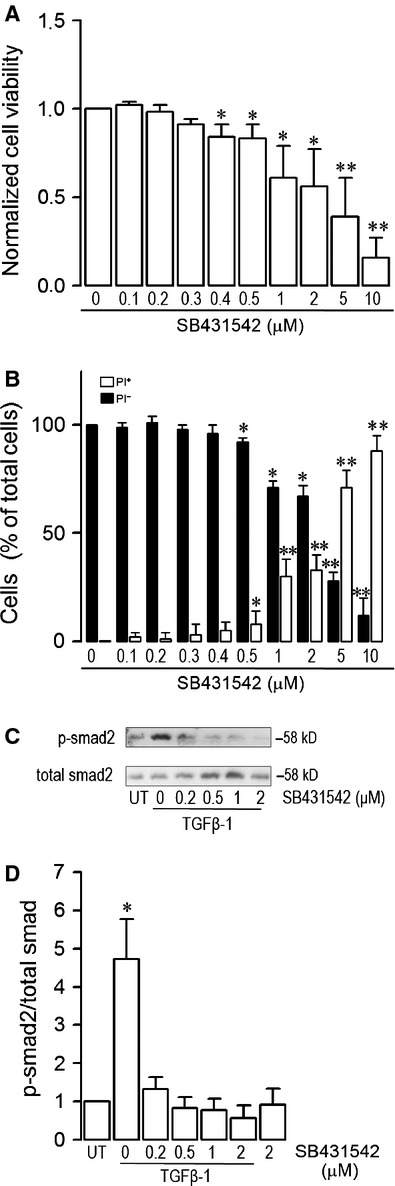
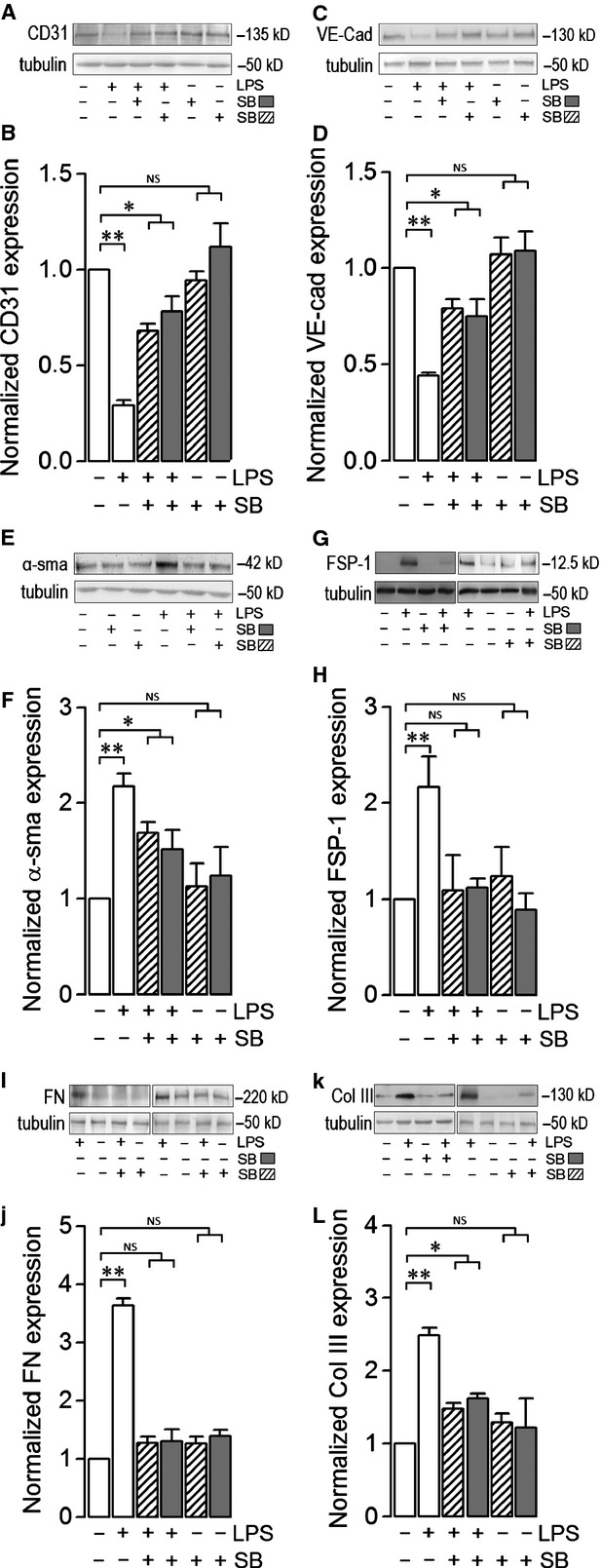
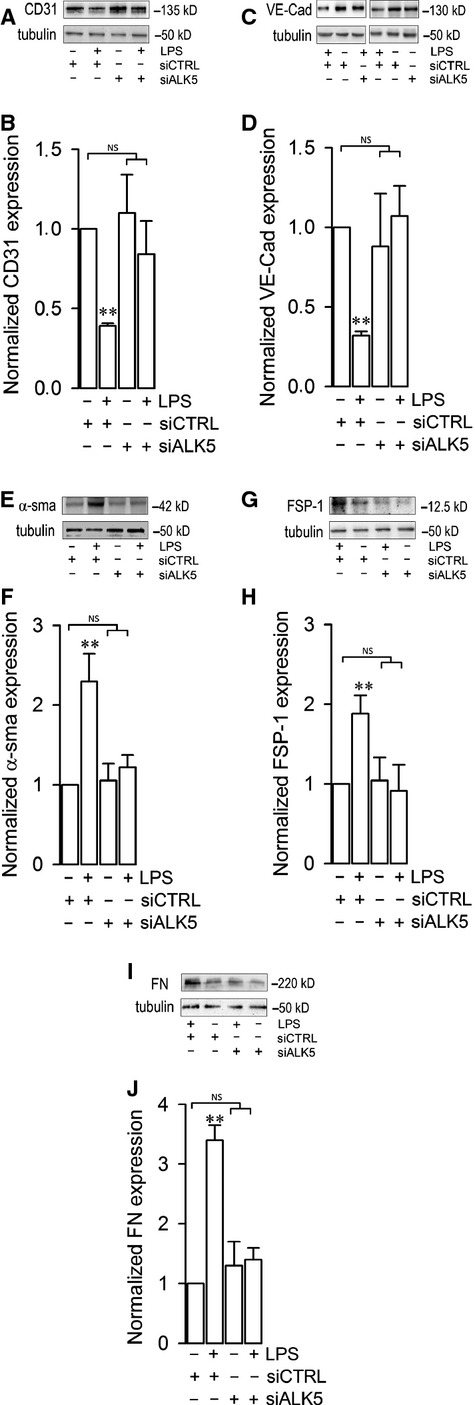
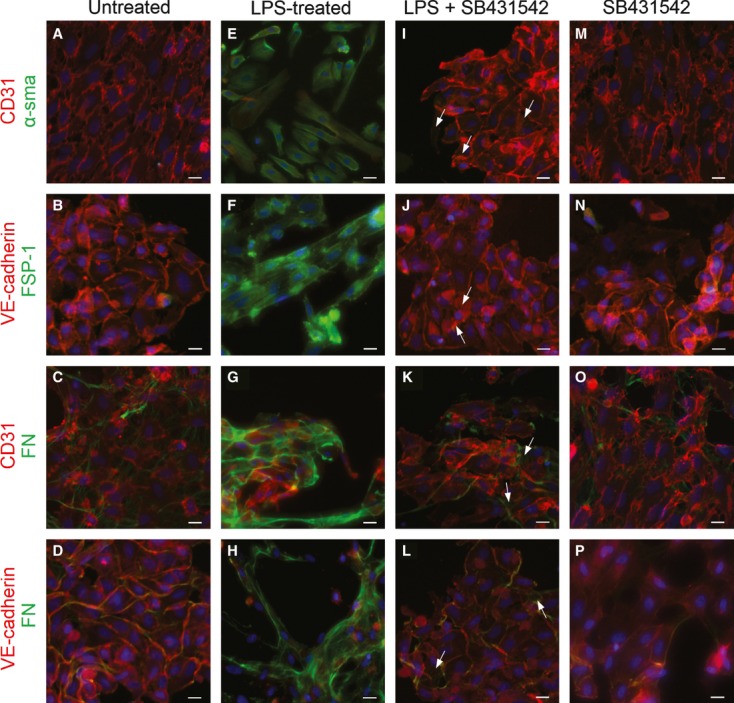
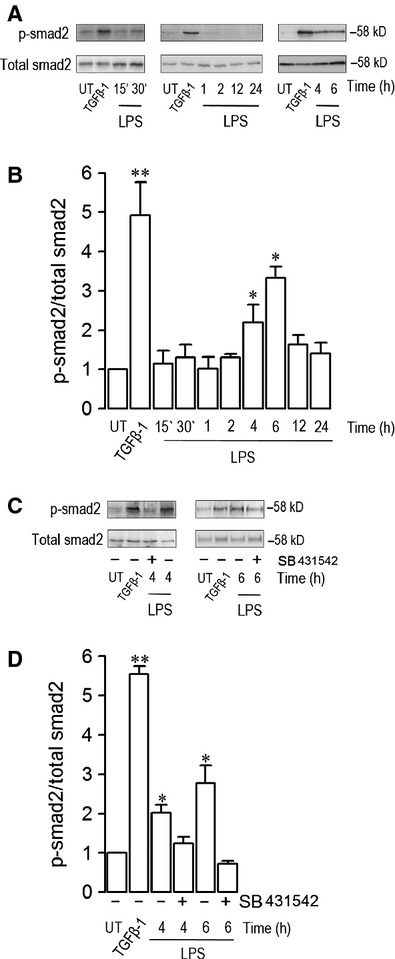
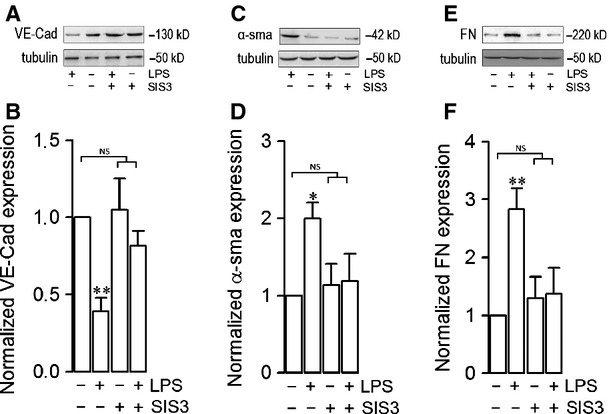
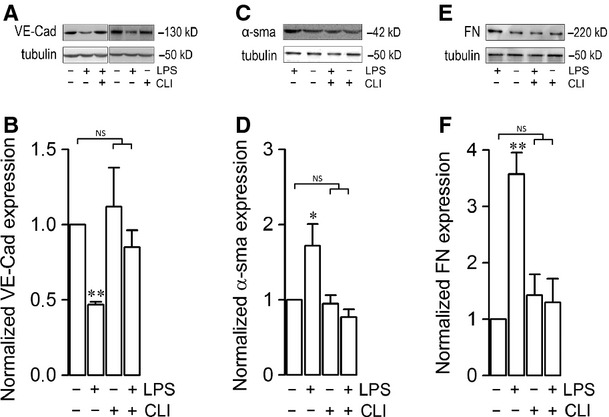
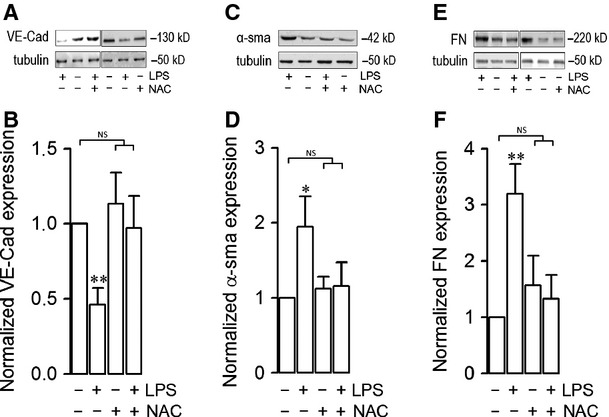
Endothelial dysfunction is crucial in endotoxaemia-derived sepsis syndrome pathogenesis. It is well accepted that lipopolysaccharide (LPS) induces endothelial dysfunction through immune system activation. However, LPS can also directly generate actions in endothelial cells (ECs) in the absence of participation by immune cells. Although interactions between LPS and ECs evoke endothelial death, a significant portion of ECs are resistant to LPS challenge. However, the mechanism that confers endothelial resistance to LPS is not known. LPS-resistant ECs exhibit a fibroblast-like morphology, suggesting that these ECs enter a fibrotic programme in response to LPS. Thus, our aim was to investigate whether LPS is able to induce endothelial fibrosis in the absence of immune cells and explore the underlying mechanism. Using primary cultures of ECs and culturing intact blood vessels, we demonstrated that LPS is a crucial factor to induce endothelial fibrosis. We demonstrated that LPS was able and sufficient to promote endothelial fibrosis, in the absence of immune cells through an activin receptor-like kinase 5 (ALK5) activity-dependent mechanism. LPS-challenged ECs showed an up-regulation of both fibroblast-specific protein expression and extracellular matrix proteins secretion, as well as a down-regulation of endothelial markers. These results demonstrate that LPS is a crucial factor in inducing endothelial fibrosis in the absence of immune cells through an ALK5-dependent mechanism. It is noteworthy that LPS-induced endothelial fibrosis perpetuates endothelial dysfunction as a maladaptive process rather than a survival mechanism for protection against LPS. These findings are useful in improving current treatment against endotoxaemia-derived sepsis syndrome and other inflammatory diseases.
内皮功能障碍在内毒素血症衍生的败血症综合征发病机制中至关重要。人们普遍认为,脂多糖(LPS)通过免疫系统激活诱导内皮功能障碍。然而,LPS 也可以在没有免疫细胞参与的情况下直接在血管内皮细胞(EC)中发挥作用。尽管 LPS 与 EC 之间的相互作用会引发内皮细胞死亡,但相当一部分 EC 对 LPS 挑战具有抗性。然而,赋予内皮细胞对 LPS 抗性的机制尚不清楚。对 LPS 具有抗性的 EC 表现出成纤维细胞样形态,表明这些 EC 在 LPS 刺激下进入纤维化程序。因此,我们的目的是研究 LPS 是否能够在没有免疫细胞的情况下诱导内皮纤维化,并探讨其潜在机制。通过原代 EC 培养和完整血管培养,我们证明 LPS 是诱导内皮纤维化的关键因素。我们证明,LPS 能够通过激活素受体样激酶 5(ALK5)活性依赖性机制,在没有免疫细胞的情况下,充分促进内皮纤维化。LPS 刺激的 EC 表现出成纤维细胞特异性蛋白表达和细胞外基质蛋白分泌的上调,以及内皮标志物的下调。这些结果表明,LPS 是在没有免疫细胞的情况下通过 ALK5 依赖性机制诱导内皮纤维化的关键因素。值得注意的是,LPS 诱导的内皮纤维化作为一种适应不良的过程而不是保护免受 LPS 侵害的生存机制,会导致内皮功能障碍持续存在。这些发现有助于改善针对内毒素血症衍生的败血症综合征和其他炎症性疾病的当前治疗方法。