Comprehensive Cancer Center and Department of Internal Medicine, University of Michigan, 1500 East Medical Center Drive, Ann Arbor, Michigan 48109, USA.
J Am Chem Soc. 2013 May 15;135(19):7223-34. doi: 10.1021/ja3125417. Epub 2013 May 3.
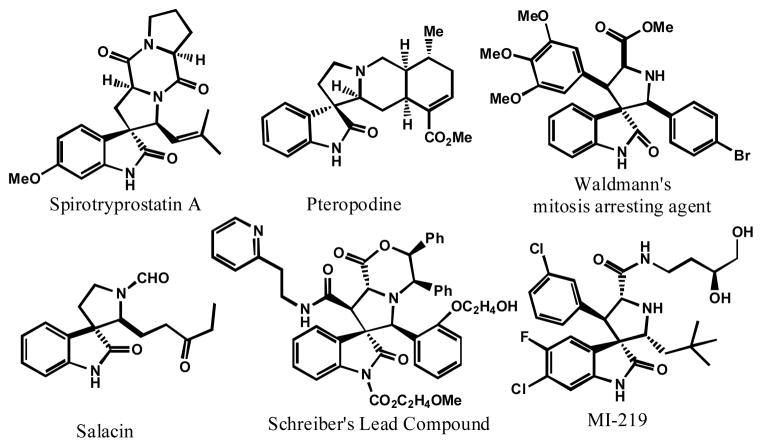
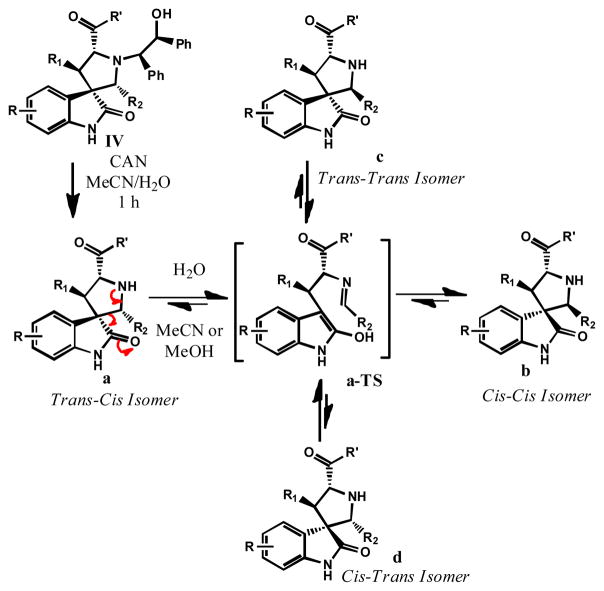
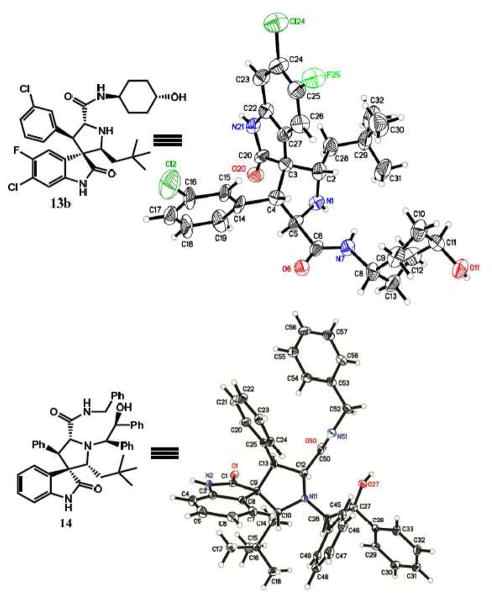
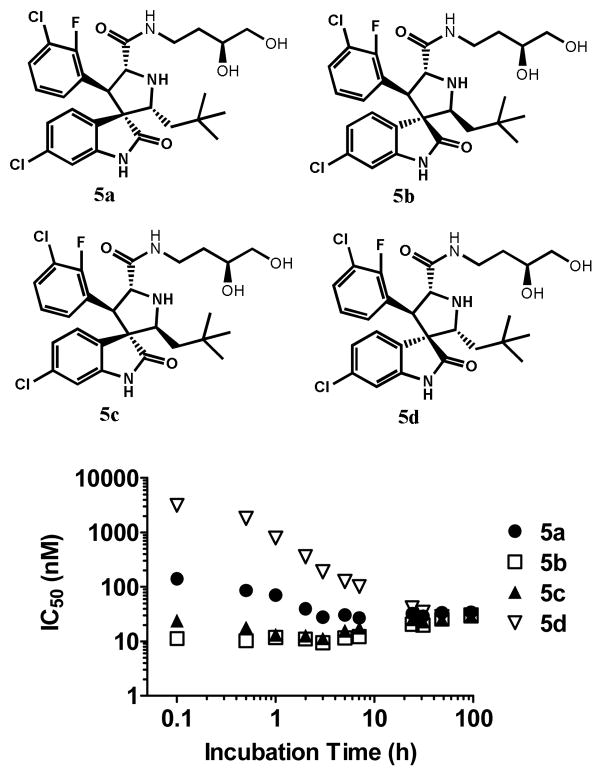
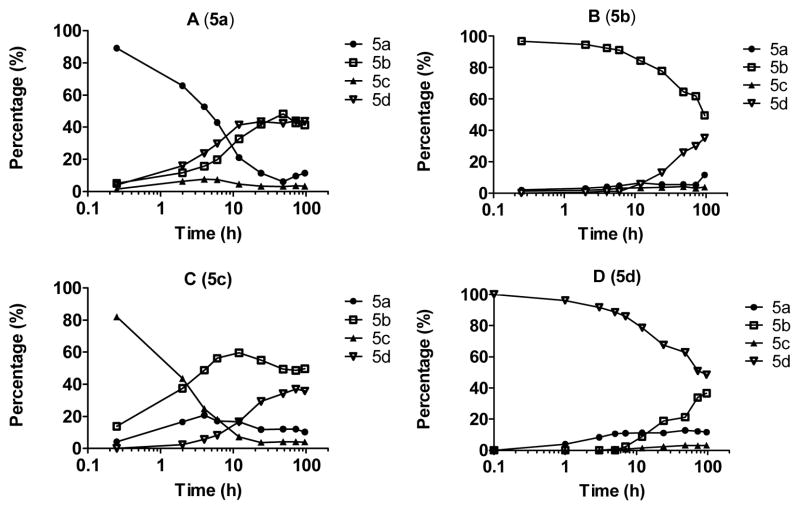
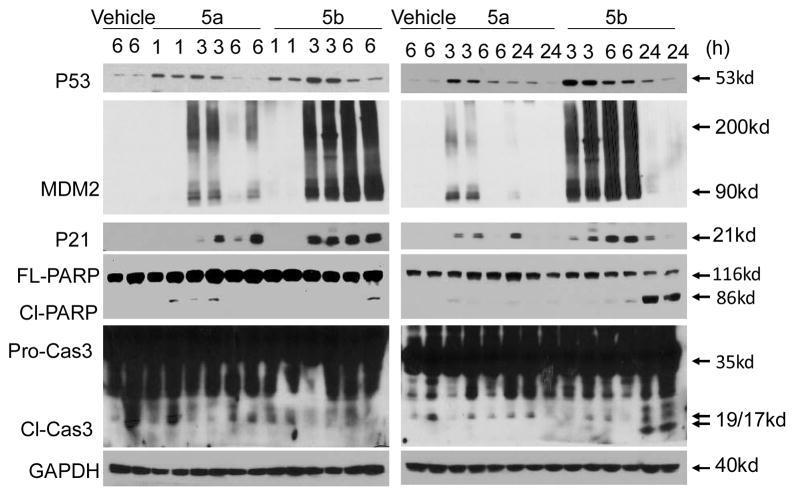
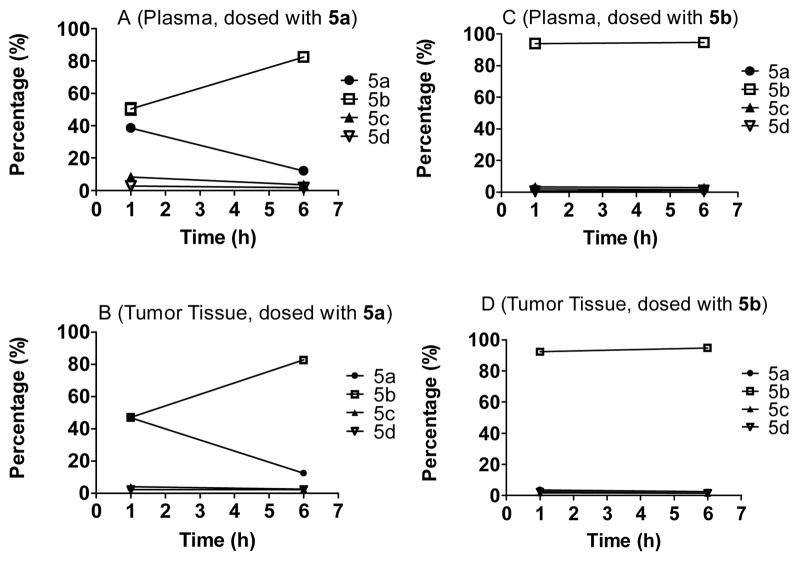
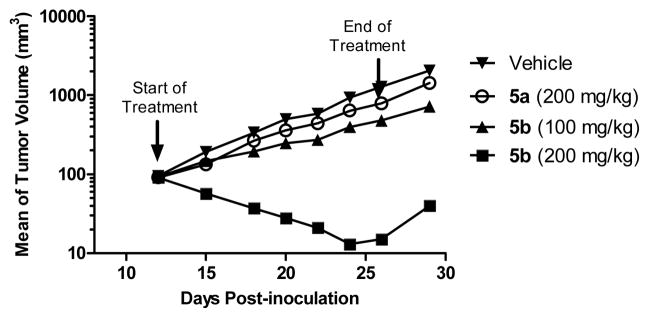
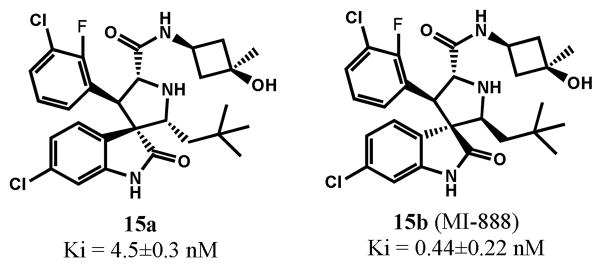
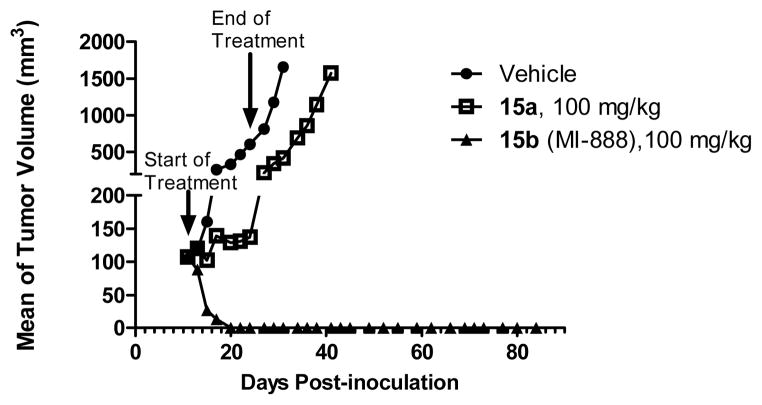
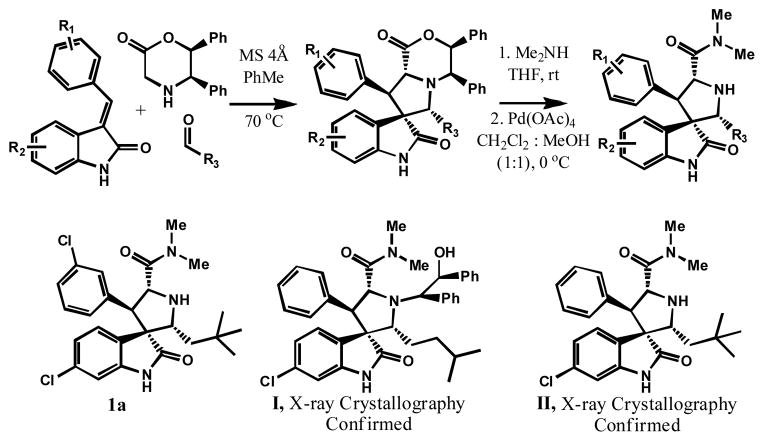
Small-molecule inhibitors that block the MDM2-p53 protein-protein interaction (MDM2 inhibitors) are being intensely pursued as a new therapeutic strategy for cancer treatment. We previously published a series of spirooxindole-containing compounds as a new class of MDM2 small-molecule inhibitors. We report herein a reversible ring-opening-cyclization reaction for some of these spirooxindoles, which affords four diastereomers from a single compound. Our biochemical binding data showed that the stereochemistry in this class of compounds has a major effect on their binding affinities to MDM2, with >100-fold difference between the most potent and the least potent stereoisomers. Our study has led to the identification of a set of highly potent MDM2 inhibitors with a stereochemistry that is different from that of our previously reported compounds. The most potent compound (MI-888) binds to MDM2 with a Ki value of 0.44 nM and achieves complete and long-lasting tumor regression in an animal model of human cancer.
小分子抑制剂可阻断 MDM2-p53 蛋白-蛋白相互作用(MDM2 抑制剂),作为癌症治疗的一种新的治疗策略正受到广泛关注。我们之前发表了一系列含有螺环吲哚的化合物,将其作为一类新的 MDM2 小分子抑制剂。本文报道了其中一些螺环吲哚的可还原的开环-环化反应,该反应从单个化合物中提供了四个非对映异构体。我们的生化结合数据表明,此类化合物的立体化学对其与 MDM2 的结合亲和力有重大影响,最强和最弱的立体异构体之间的差异超过 100 倍。我们的研究导致了一组具有不同于我们之前报道的化合物的立体化学的高度有效的 MDM2 抑制剂的鉴定。最有效的化合物(MI-888)与 MDM2 的 Ki 值为 0.44 nM,并在人类癌症动物模型中实现完全和持久的肿瘤消退。