Structural Genomics Consortium, Nuffield Department of Clinical Medicine, University of Oxford, Old Road Campus Research Building, Roosevelt Drive, Oxford, OX3 7DQ, UK.
J Med Chem. 2013 Jun 13;56(11):4413-21. doi: 10.1021/jm4000837. Epub 2013 May 17.
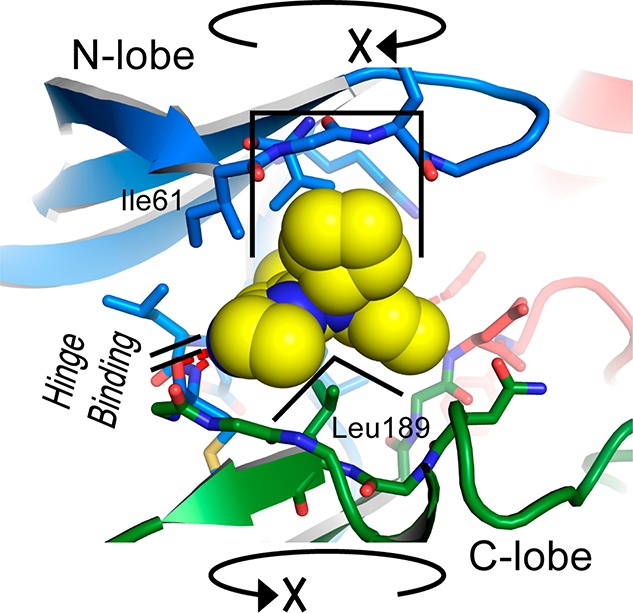
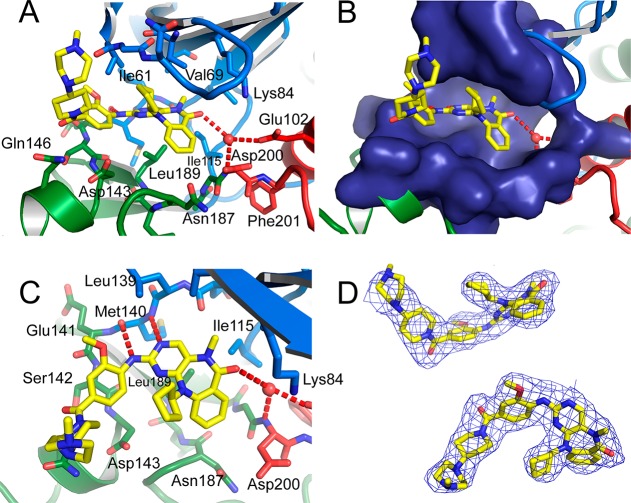
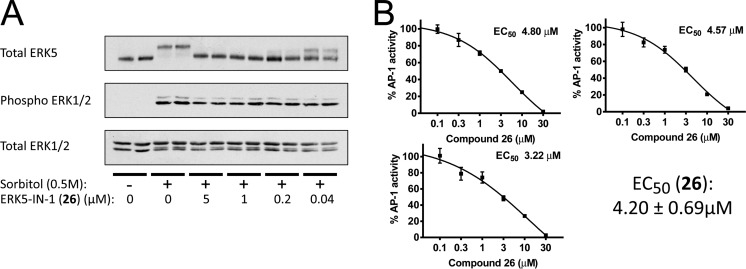
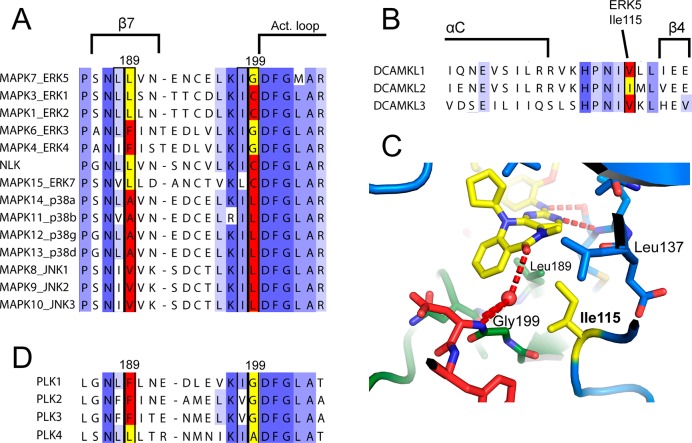
The protein kinase ERK5 (MAPK7) is an emerging drug target for a variety of indications, in particular for cancer where it plays a key role mediating cell proliferation, survival, epithelial-mesenchymal transition, and angiogenesis. To date, no three-dimensional structure has been published that would allow rational design of inhibitors. To address this, we determined the X-ray crystal structure of the human ERK5 kinase domain in complex with a highly specific benzo[e]pyrimido[5,4-b]diazepine-6(11H)-one inhibitor. The structure reveals that specific residue differences in the ATP-binding site, compared to the related ERKs p38s and JNKs, allow for the development of ERK5-specific inhibitors. The selectivity of previously observed ERK5 inhibitors can also be rationalized using this structure, which provides a template for future development of inhibitors with potential for treatment of disease.
丝裂原活化蛋白激酶 ERK5(MAPK7)是多种适应症的新兴药物靶点,特别是在癌症中,它在介导细胞增殖、存活、上皮-间充质转化和血管生成方面发挥着关键作用。迄今为止,尚无发表的三维结构能够允许进行抑制剂的合理设计。为了解决这个问题,我们测定了与人 ERK5 激酶结构域与高度特异性苯并[e]嘧啶并[5,4-b]二氮杂酮-6(11H)-酮抑制剂复合物的 X 射线晶体结构。该结构表明,与相关的 ERKs p38 和 JNKs 相比,ATP 结合位点的特定残基差异允许开发 ERK5 特异性抑制剂。使用该结构也可以合理地解释先前观察到的 ERK5 抑制剂的选择性,为具有治疗疾病潜力的抑制剂的未来发展提供了模板。