University of Manchester, Manchester Academic Health Science Centre, Salford Royal NHS Foundation Trust, Salford, UK.
Mol Genet Metab. 2013 Sep-Oct;110(1-2):54-64. doi: 10.1016/j.ymgme.2013.04.002. Epub 2013 Apr 10.
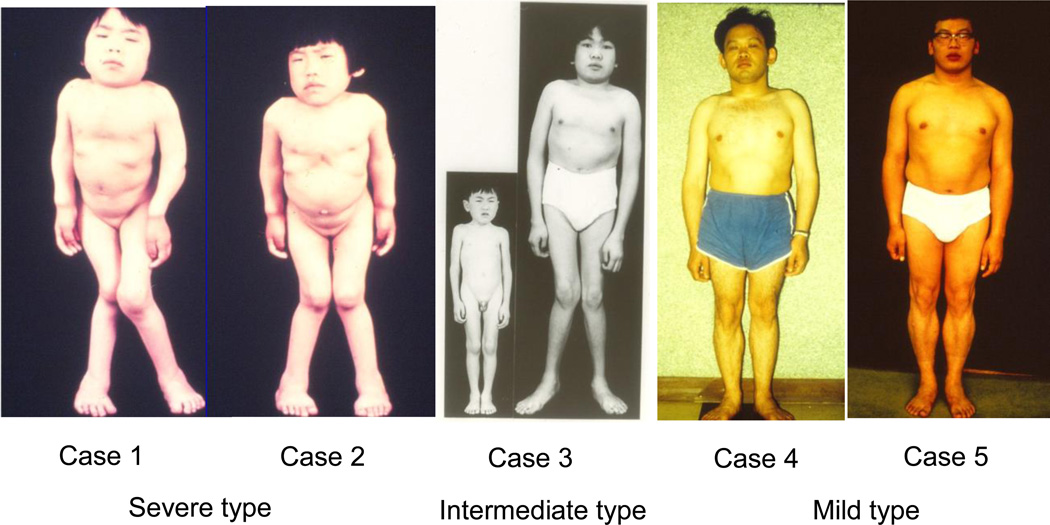
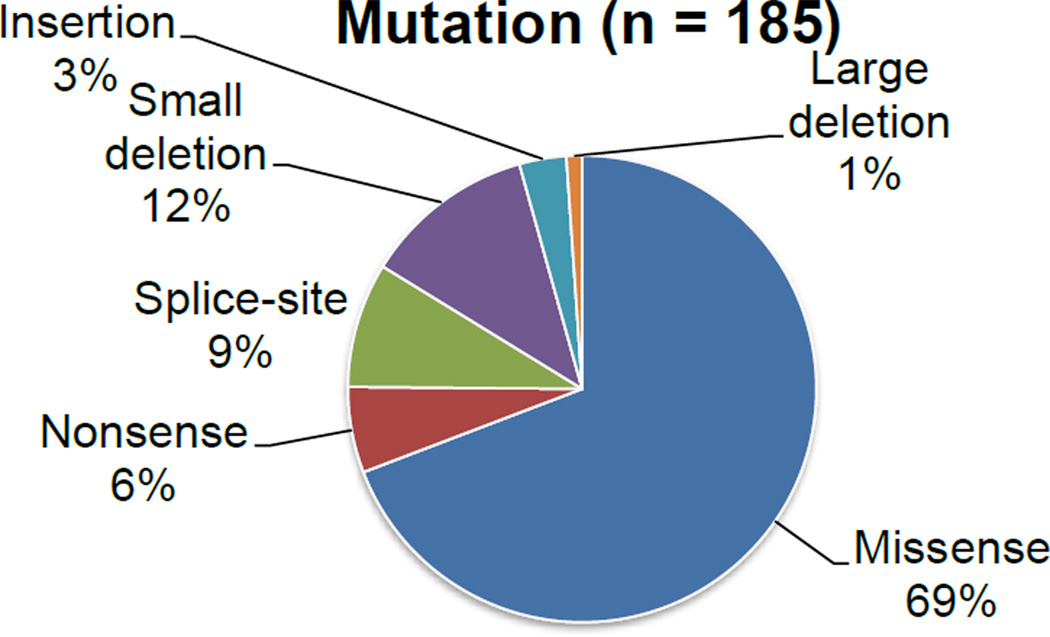
Mucopolysaccharidosis type IVA (MPS IVA) was described in 1929 by Luis Morquio from Uruguay and James Brailsford from England, and was later found as an autosomal recessive lysosomal storage disease. MPS IVA is caused by mutations in the gene encoding the enzyme, N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Reduced GALNS activity results in impaired catabolism of two glycosaminoglycans (GAGs), chondroitin-6-sulfate (C6S) and keratan sulfate (KS). Clinical presentations of MPS IVA reflect a spectrum of progression from a severe "classical" phenotype to a mild "attenuated" phenotype. More than 180 different mutations have been identified in the GALNS gene, which likely explains the phenotypic heterogeneity of the disorder. Accumulation of C6S and KS manifests predominantly as short stature and skeletal dysplasia (dysostosis multiplex), including atlantoaxial instability and cervical cord compression. However, abnormalities in the visual, auditory, cardiovascular, and respiratory systems can also affect individuals with MPS IVA. Diagnosis is typically based on clinical examination, skeletal radiographs, urinary GAG, and enzymatic activity of GALNS in blood cells or fibroblasts. Deficiency of GALNS activity is a common assessment for the laboratory diagnosis of MPS IVA; however, with recently increased availability, gene sequencing for MPS IVA is often used to confirm enzyme results. As multiple clinical presentations are observed, diagnosis of MPS IVA may require multi-system considerations. This review provides a history of defining MPS IVA and how the understanding of the disease manifestations has changed over time. A summary of the accumulated knowledge is presented, including information from the International Morquio Registry. The classical phenotype is contrasted with attenuated cases, which are now being recognized and diagnosed more frequently. Laboratory based diagnoses of MPS IVA are also discussed.
黏多糖贮积症 IVA 型(MPS IVA)于 1929 年由来自乌拉圭的 Luis Morquio 和来自英国的 James Brailsford 首次描述,后来被发现是一种常染色体隐性溶酶体贮积病。MPS IVA 是由编码酶 N-乙酰半乳糖胺-6-硫酸酯硫酸酯酶(GALNS)的基因突变引起的。GALNS 活性降低导致两种糖胺聚糖(GAGs),即软骨素-6-硫酸酯(C6S)和硫酸角质素(KS)的分解代谢受损。MPS IVA 的临床表现反映了从严重的“经典”表型到轻度“衰减”表型的进展范围。在 GALNS 基因中已经发现了超过 180 种不同的突变,这可能解释了该疾病表型的异质性。C6S 和 KS 的积累主要表现为身材矮小和骨骼发育不良(多发性骨发育不良),包括寰枢椎不稳定和颈脊髓压迫。然而,视觉、听觉、心血管和呼吸系统的异常也会影响 MPS IVA 患者。诊断通常基于临床检查、骨骼 X 光片、尿 GAG 和血液细胞或成纤维细胞中 GALNS 的酶活性。GALNS 活性缺乏是 MPS IVA 实验室诊断的常用评估方法;然而,随着基因测序技术的普及,MPS IVA 的基因测序通常用于确认酶结果。由于观察到多种临床表现,因此 MPS IVA 的诊断可能需要多系统考虑。本文回顾了定义 MPS IVA 的历史以及随着时间的推移对疾病表现的理解的变化。本文总结了积累的知识,包括来自国际莫尔奎奥登记处的信息。经典表型与衰减病例形成对比,现在越来越多地认识到并诊断出衰减病例。还讨论了基于实验室的 MPS IVA 诊断。