Centre for Molecular and Biomolecular Informatics, Radboud University Medical Centre, Nijmegen, The Netherlands.
PLoS One. 2013 May 10;8(5):e63523. doi: 10.1371/journal.pone.0063523. Print 2013.
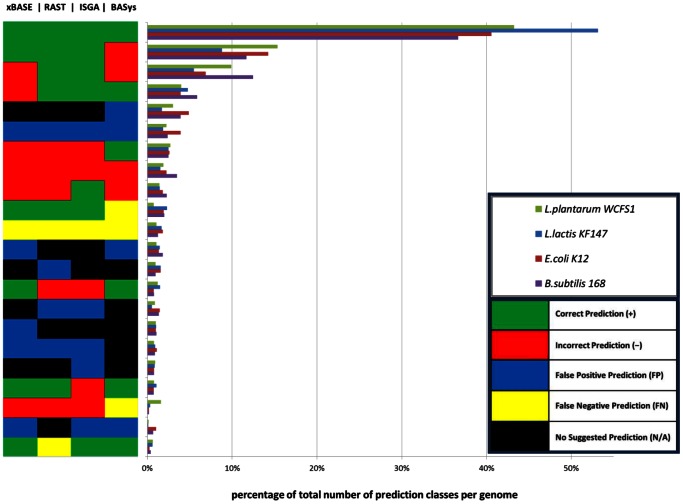
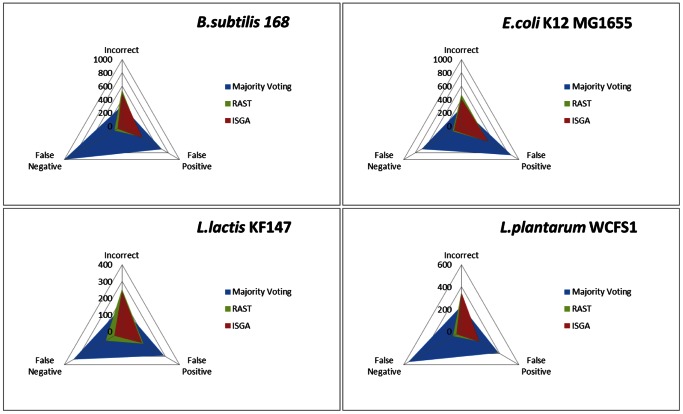
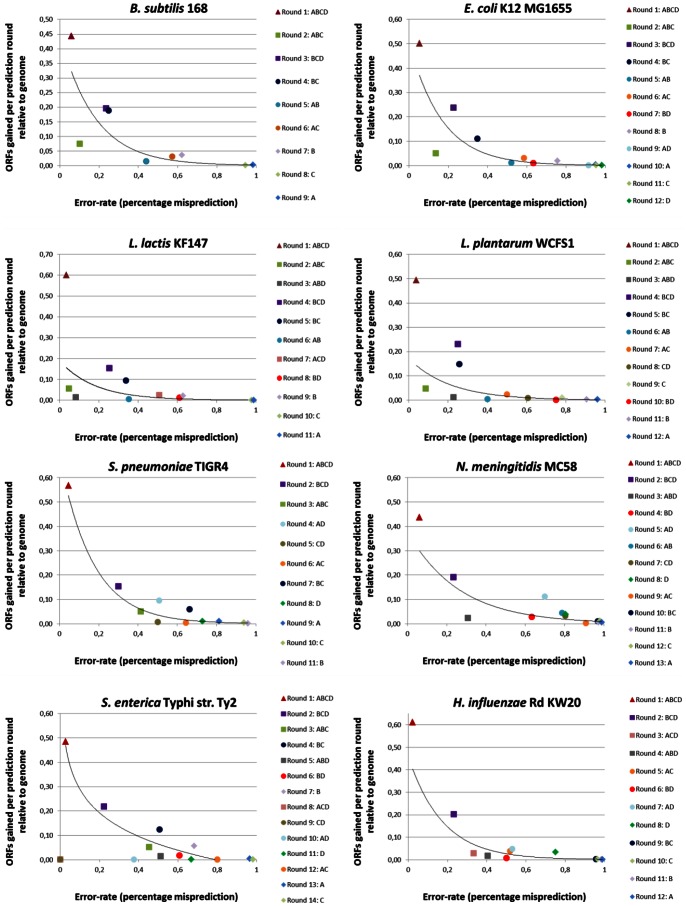
Nowadays, prokaryotic genomes are sequenced faster than the capacity to manually curate gene annotations. Automated genome annotation engines provide users a straight-forward and complete solution for predicting ORF coordinates and function. For many labs, the use of AGEs is therefore essential to decrease the time necessary for annotating a given prokaryotic genome. However, it is not uncommon for AGEs to provide different and sometimes conflicting predictions. Combining multiple AGEs might allow for more accurate predictions. Here we analyzed the ab initio open reading frame (ORF) calling performance of different AGEs based on curated genome annotations of eight strains from different bacterial species with GC% ranging from 35-52%. We present a case study which demonstrates a novel way of comparative genome annotation, using combinations of AGEs in a pre-defined order (or path) to predict ORF start codons. The order of AGE combinations is from high to low specificity, where the specificity is based on the eight genome annotations. For each AGE combination we are able to derive a so-called projected confidence value, which is the average specificity of ORF start codon prediction based on the eight genomes. The projected confidence enables estimating likeliness of a correct prediction for a particular ORF start codon by a particular AGE combination, pinpointing ORFs notoriously difficult to predict start codons. We correctly predict start codons for 90.5±4.8% of the genes in a genome (based on the eight genomes) with an accuracy of 81.1±7.6%. Our consensus-path methodology allows a marked improvement over majority voting (9.7±4.4%) and with an optimal path ORF start prediction sensitivity is gained while maintaining a high specificity.
如今,原核基因组的测序速度快于手动基因注释的能力。自动化基因组注释引擎为用户提供了一种直接而完整的解决方案,用于预测 ORF 坐标和功能。因此,对于许多实验室来说,使用 AGE 对于减少注释给定原核基因组所需的时间是必不可少的。然而,AGE 提供不同的、有时甚至相互冲突的预测并不罕见。组合多个 AGE 可能会允许更准确的预测。在这里,我们根据来自不同细菌物种的 8 个菌株的经过 curated 的基因组注释,分析了不同 AGE 的从头开始的开放阅读框 (ORF) 调用性能,这些菌株的 GC% 范围从 35-52%。我们提出了一个案例研究,展示了一种使用预定义顺序(或路径)组合 AGE 来预测 ORF 起始密码子的比较基因组注释的新方法。AGE 组合的顺序是从高特异性到低特异性,特异性是基于这 8 个基因组注释的。对于每个 AGE 组合,我们都能够得出所谓的投影置信值,它是基于 8 个基因组的 ORF 起始密码子预测的平均特异性。投影置信度使我们能够估计特定 AGE 组合对特定 ORF 起始密码子的正确预测的可能性,从而确定那些难以预测起始密码子的 ORF。我们能够正确预测 90.5±4.8%(基于 8 个基因组)的基因的起始密码子,准确率为 81.1±7.6%。我们的共识路径方法与多数投票(9.7±4.4%)相比有了显著的改进,并且通过最优路径,在保持高特异性的同时获得了 ORF 起始预测的敏感性。