Genomic Signal Processing Laboratory, Electrical and Computer Engineering, Drexel University, Philadelphia, PA 19104, USA.
BMC Bioinformatics. 2011 Jan 13;12:20. doi: 10.1186/1471-2105-12-20.
Traditional gene annotation methods rely on characteristics that may not be available in short reads generated from next generation technology, resulting in suboptimal performance for metagenomic (environmental) samples. Therefore, in recent years, new programs have been developed that optimize performance on short reads. In this work, we benchmark three metagenomic gene prediction programs and combine their predictions to improve metagenomic read gene annotation.
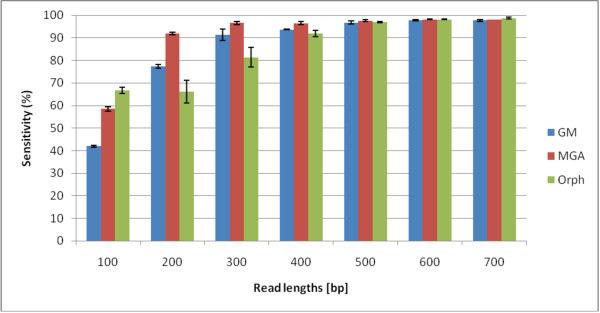
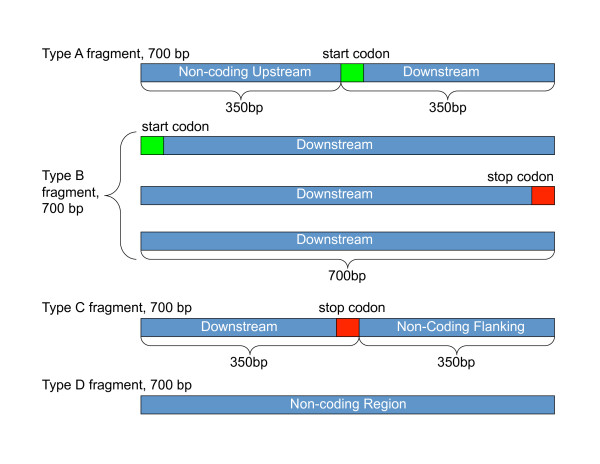
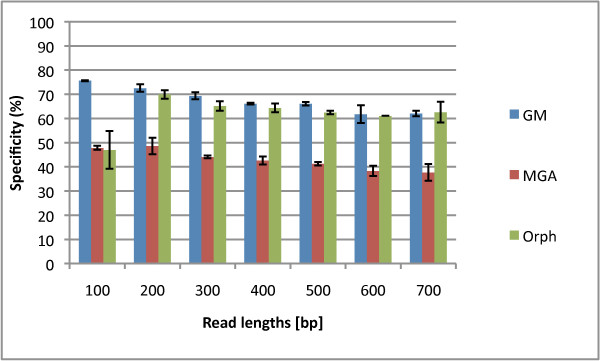
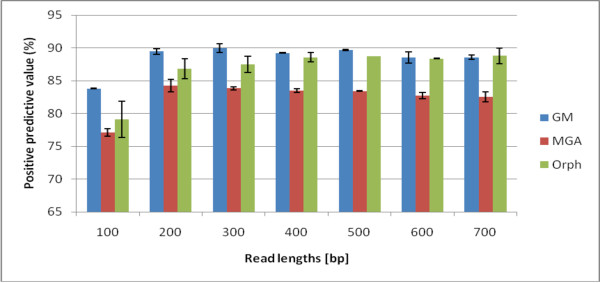
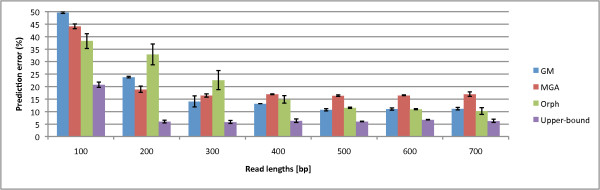
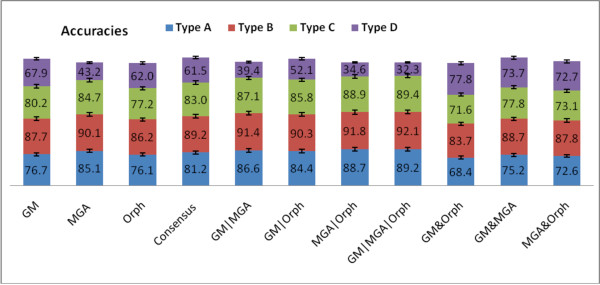
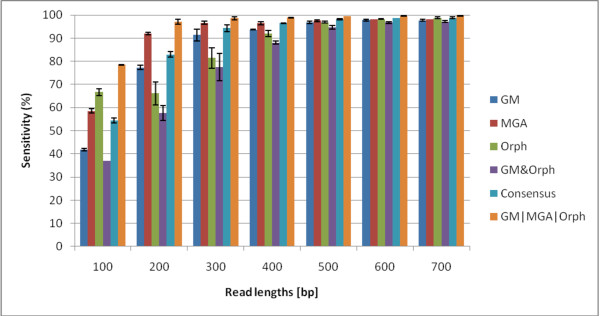
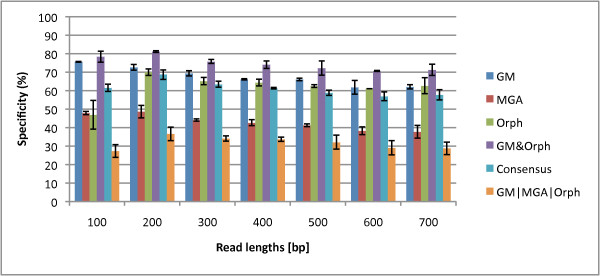
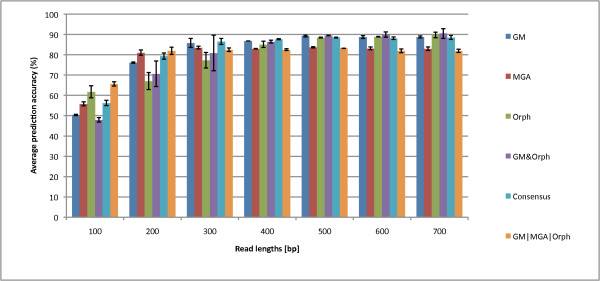
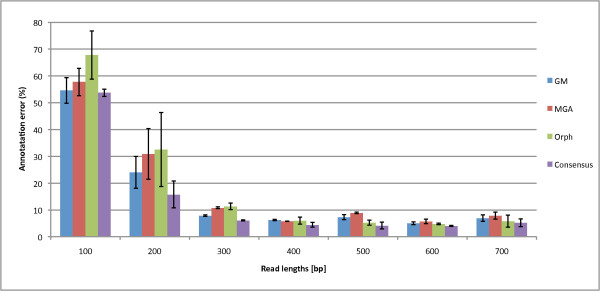
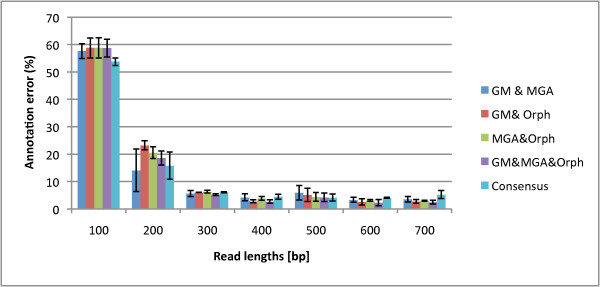
We not only analyze the programs' performance at different read-lengths like similar studies, but also separate different types of reads, including intra- and intergenic regions, for analysis. The main deficiencies are in the algorithms' ability to predict non-coding regions and gene edges, resulting in more false-positives and false-negatives than desired. In fact, the specificities of the algorithms are notably worse than the sensitivities. By combining the programs' predictions, we show significant improvement in specificity at minimal cost to sensitivity, resulting in 4% improvement in accuracy for 100 bp reads with ~1% improvement in accuracy for 200 bp reads and above. To correctly annotate the start and stop of the genes, we find that a consensus of all the predictors performs best for shorter read lengths while a unanimous agreement is better for longer read lengths, boosting annotation accuracy by 1-8%. We also demonstrate use of the classifier combinations on a real dataset.
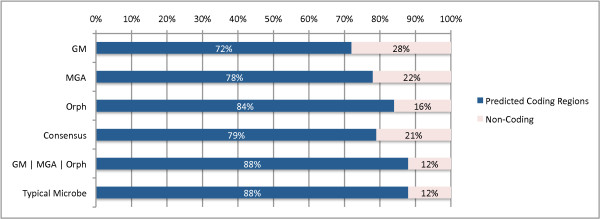
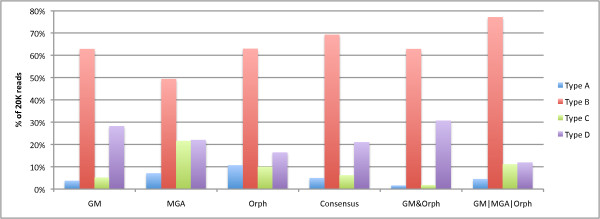
To optimize the performance for both prediction and annotation accuracies, we conclude that the consensus of all methods (or a majority vote) is the best for reads 400 bp and shorter, while using the intersection of GeneMark and Orphelia predictions is the best for reads 500 bp and longer. We demonstrate that most methods predict over 80% coding (including partially coding) reads on a real human gut sample sequenced by Illumina technology.
传统的基因注释方法依赖于可能无法在下一代技术生成的短读段中获得的特征,因此对宏基因组(环境)样本的性能并不理想。因此,近年来,开发了新的程序,这些程序针对短读段进行了性能优化。在这项工作中,我们对三种宏基因组基因预测程序进行了基准测试,并结合它们的预测来改进宏基因组读段的基因注释。
我们不仅像类似的研究一样分析了不同读段长度下程序的性能,还分别分析了不同类型的读段,包括基因内和基因间区域。主要的缺陷在于算法预测非编码区域和基因边缘的能力,导致假阳性和假阴性比预期的多。事实上,算法的特异性明显比敏感性差。通过结合程序的预测,我们在最小化敏感性成本的情况下显著提高了特异性,从而使 100bp 读段的准确性提高了 4%,200bp 读段及以上的准确性提高了约 1%。为了正确注释基因的起始和结束,我们发现对于较短的读段,所有预测器的共识表现最佳,而对于较长的读段,一致的意见更好,将注释准确性提高了 1-8%。我们还在真实数据集上演示了分类器组合的使用。
为了优化预测和注释准确性的性能,我们得出结论,对于 400bp 及更短的读段,所有方法的共识(或多数票)是最佳的,而对于 500bp 及更长的读段,使用 GeneMark 和 Orphelia 预测的交集是最佳的。我们证明,大多数方法在使用 Illumina 技术测序的真实人类肠道样本中预测了超过 80%的编码(包括部分编码)读段。