Greenfest-Allen Emily, Malik Jeffrey, Palis James, Stoeckert Christian J
Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA. stoec
BMC Syst Biol. 2013 May 15;7:38. doi: 10.1186/1752-0509-7-38.
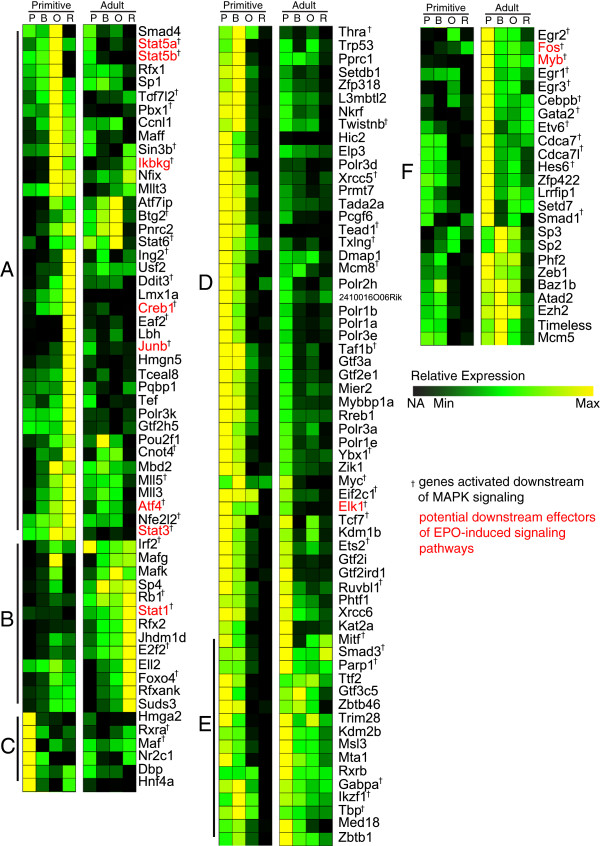
Hematopoietic ontogeny is characterized by overlapping waves of primitive, fetal definitive, and adult definitive erythroid lineages. Our aim is to identify differences in the transcriptional control of these distinct erythroid cell maturation pathways by inferring and analyzing gene-interaction networks from lineage-specific expression datasets. Inferred networks are strongly connected and do not fit a scale-free model, making it difficult to identify essential regulators using the hub-essentiality standard.
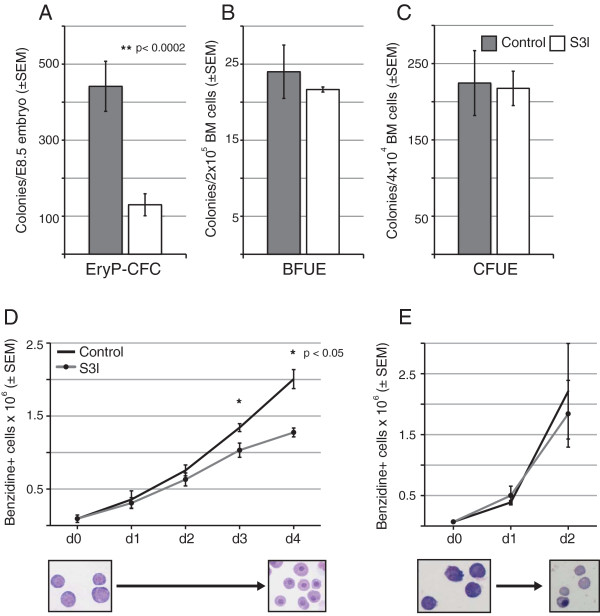
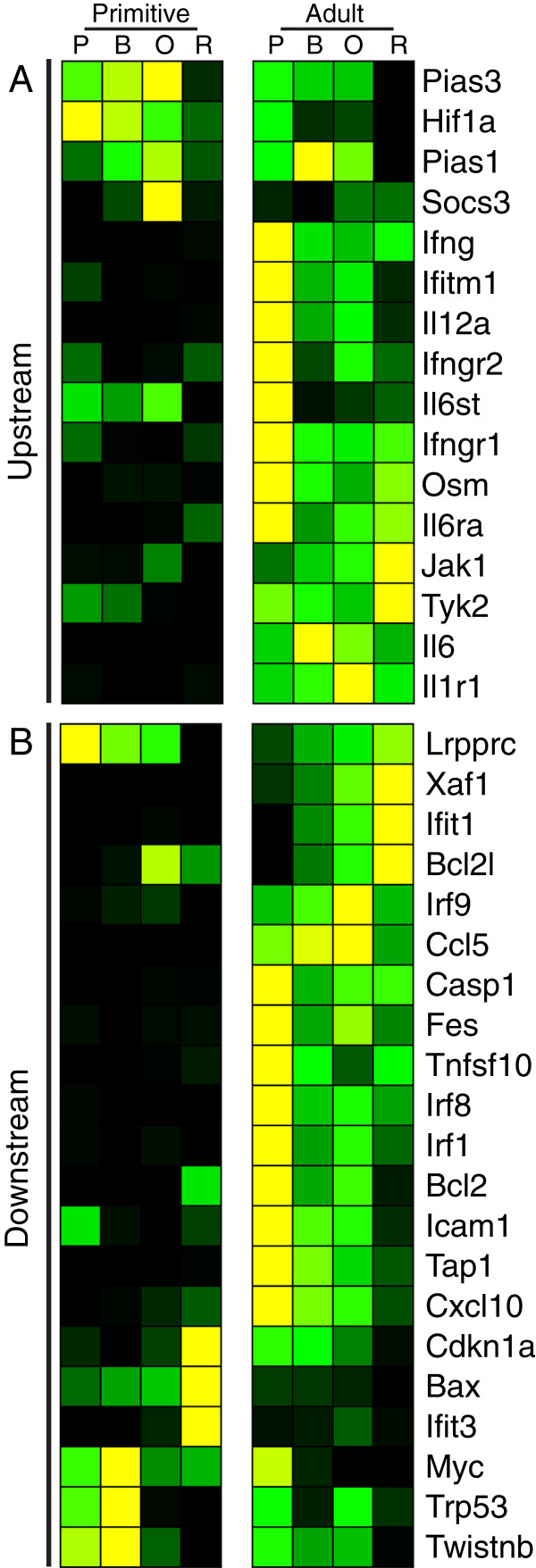
We employed a semi-supervised machine learning approach to integrate measures of network topology with expression data to score gene essentiality. The algorithm was trained and tested on the adult and fetal definitive erythroid lineages. When applied to the primitive erythroid lineage, 144 high scoring transcription factors were found to be differentially expressed between the primitive and adult definitive erythroid lineages, including all expressed STAT-family members. Differential responses of primitive and definitive erythroblasts to a Stat3 inhibitor and IFNγ in vitro supported the results of the computational analysis. Further investigation of the original expression data revealed a striking signature of Stat1-related genes in the adult definitive erythroid network. Among the potential pathways known to utilize Stat1, interferon (IFN) signaling-related genes were expressed almost exclusively within the adult definitive erythroid network.
In vitro results support the computational prediction that differential regulation and downstream effectors of STAT signaling are key factors that distinguish the transcriptional control of primitive and definitive erythroid cell maturation.
造血系统发育的特征是原始、胎儿期定型和成体期定型红系谱系的重叠浪潮。我们的目标是通过从谱系特异性表达数据集中推断和分析基因相互作用网络,来识别这些不同红系细胞成熟途径在转录调控方面的差异。推断出的网络具有强连接性,不符合无标度模型,因此难以使用枢纽基因重要性标准来识别关键调控因子。
我们采用了一种半监督机器学习方法,将网络拓扑度量与表达数据相结合,以对基因重要性进行评分。该算法在成体期和胎儿期定型红系谱系上进行了训练和测试。当应用于原始红系谱系时,发现144个高分转录因子在原始红系谱系和成体期定型红系谱系之间存在差异表达,包括所有表达的STAT家族成员。原始和成体期成红细胞在体外对Stat3抑制剂和IFNγ的不同反应支持了计算分析的结果。对原始表达数据的进一步研究揭示了成体期定型红系网络中Stat1相关基因的显著特征。在已知利用Stat1的潜在途径中,干扰素(IFN)信号相关基因几乎只在成体期定型红系网络中表达。
体外实验结果支持了计算预测,即STAT信号的差异调节和下游效应器是区分原始和成体期定型红系细胞成熟转录调控的关键因素。