Department of Neurophysiology and Neuropharmacology, Center of Physiology and Pharmacology, Medical University of Vienna, Waehringerstrasse 13a, 1090, Vienna, Austria.
Neuromolecular Med. 2013 Sep;15(3):476-92. doi: 10.1007/s12017-013-8234-1. Epub 2013 May 22.
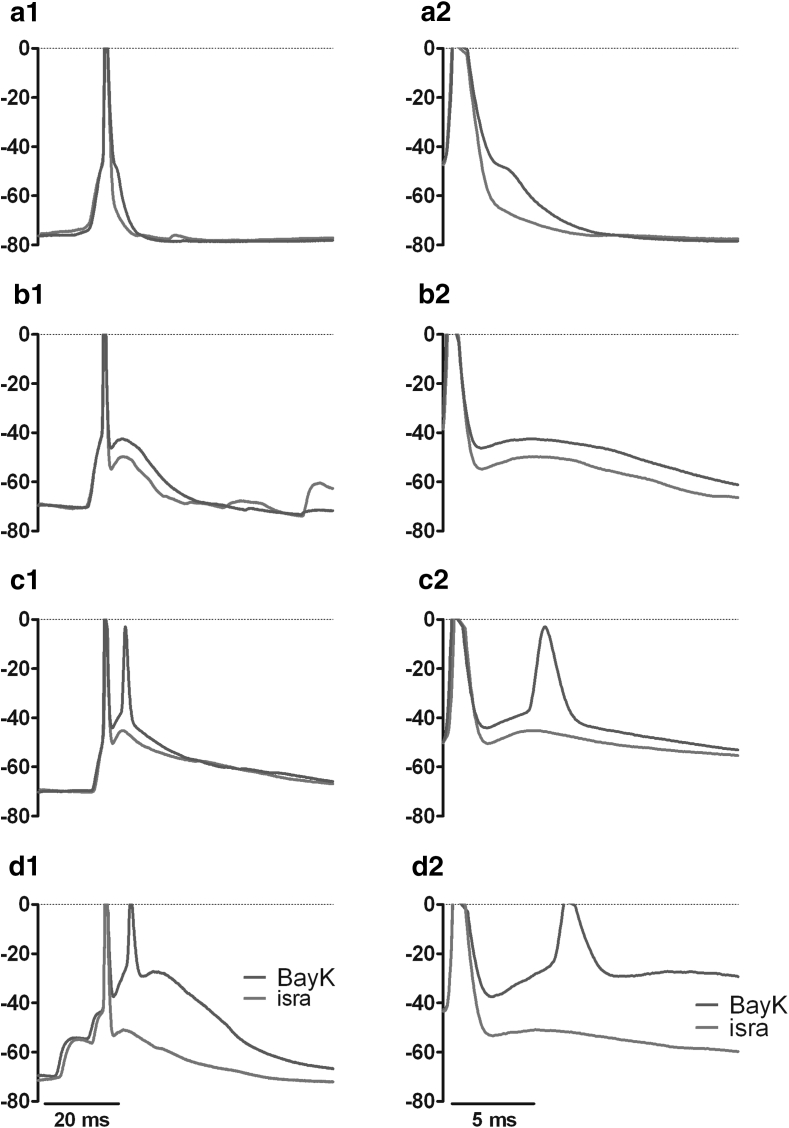
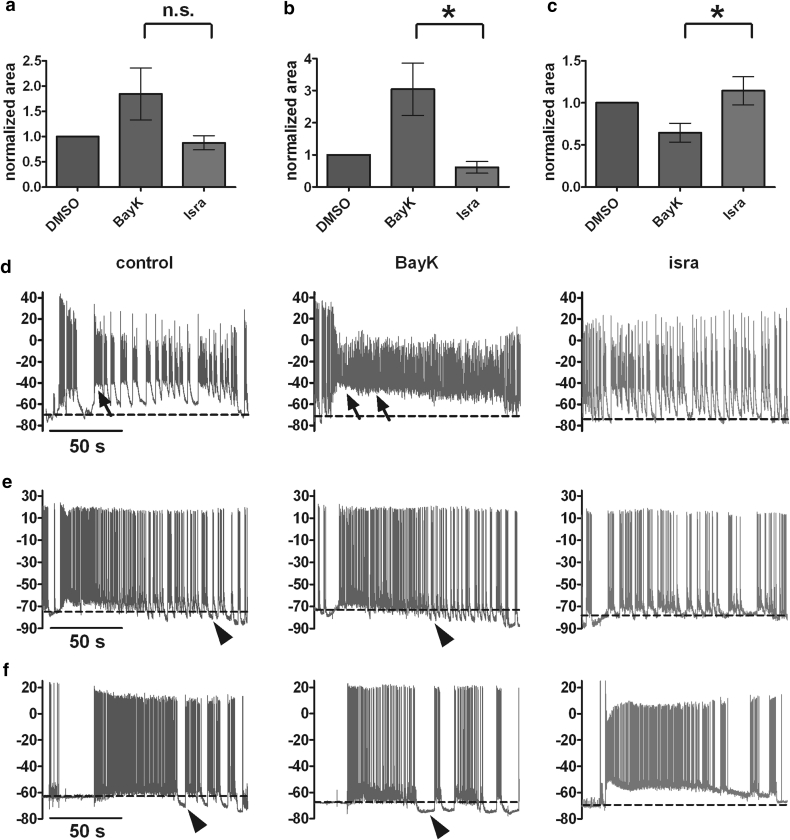
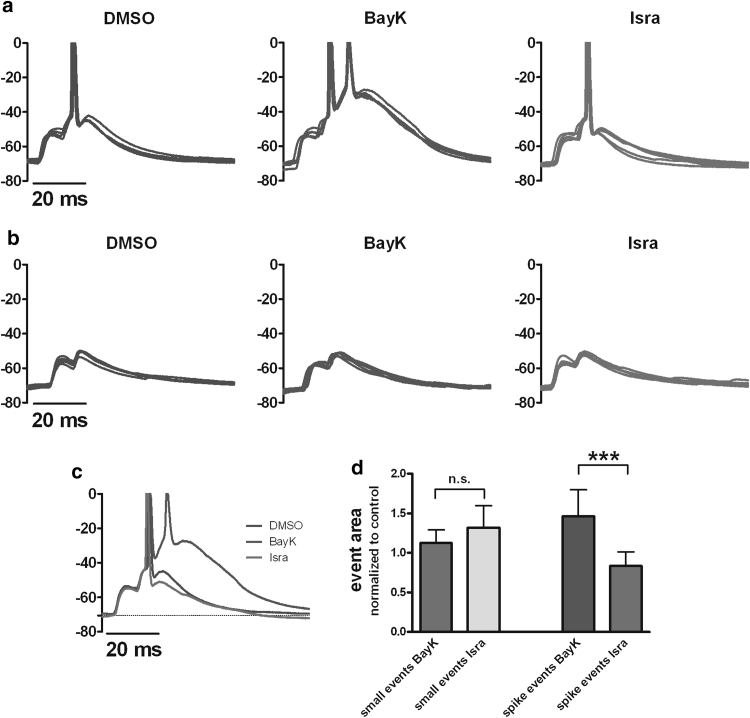
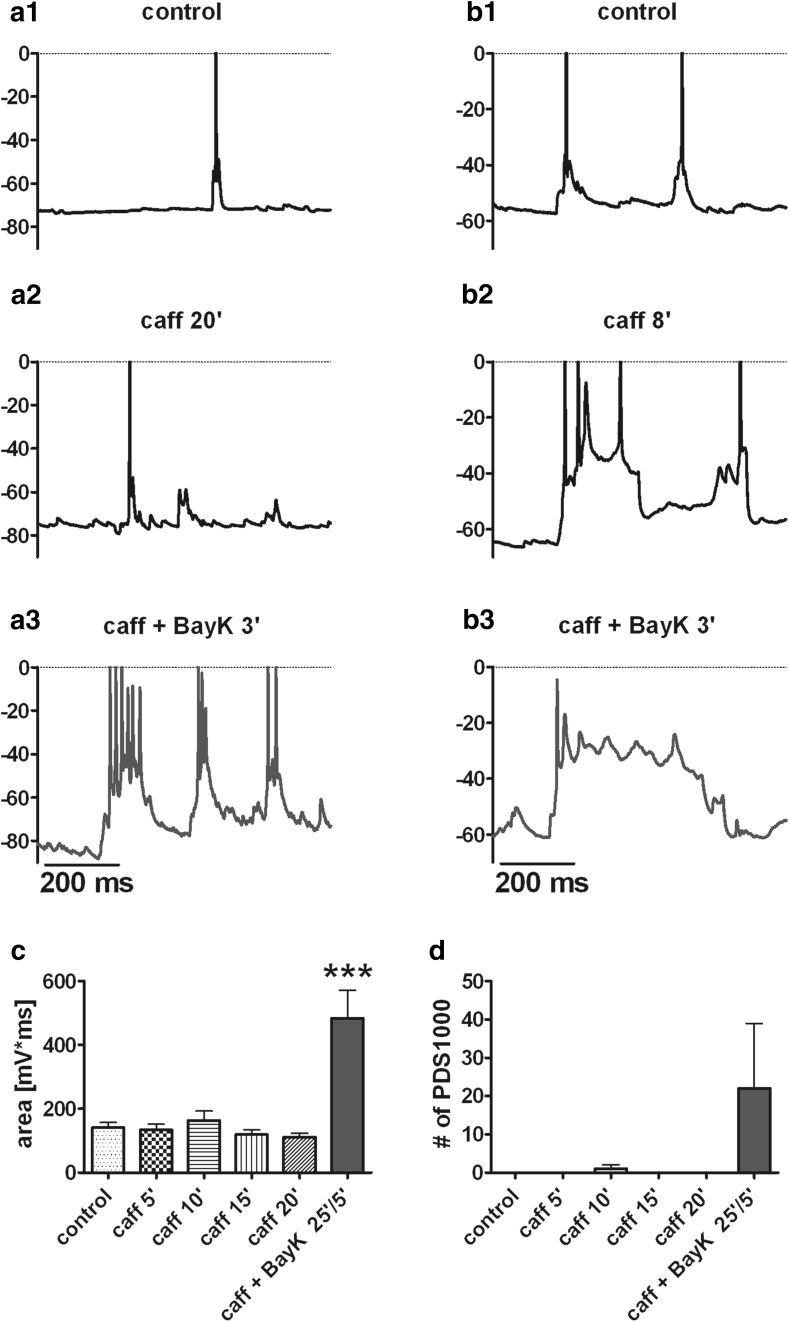
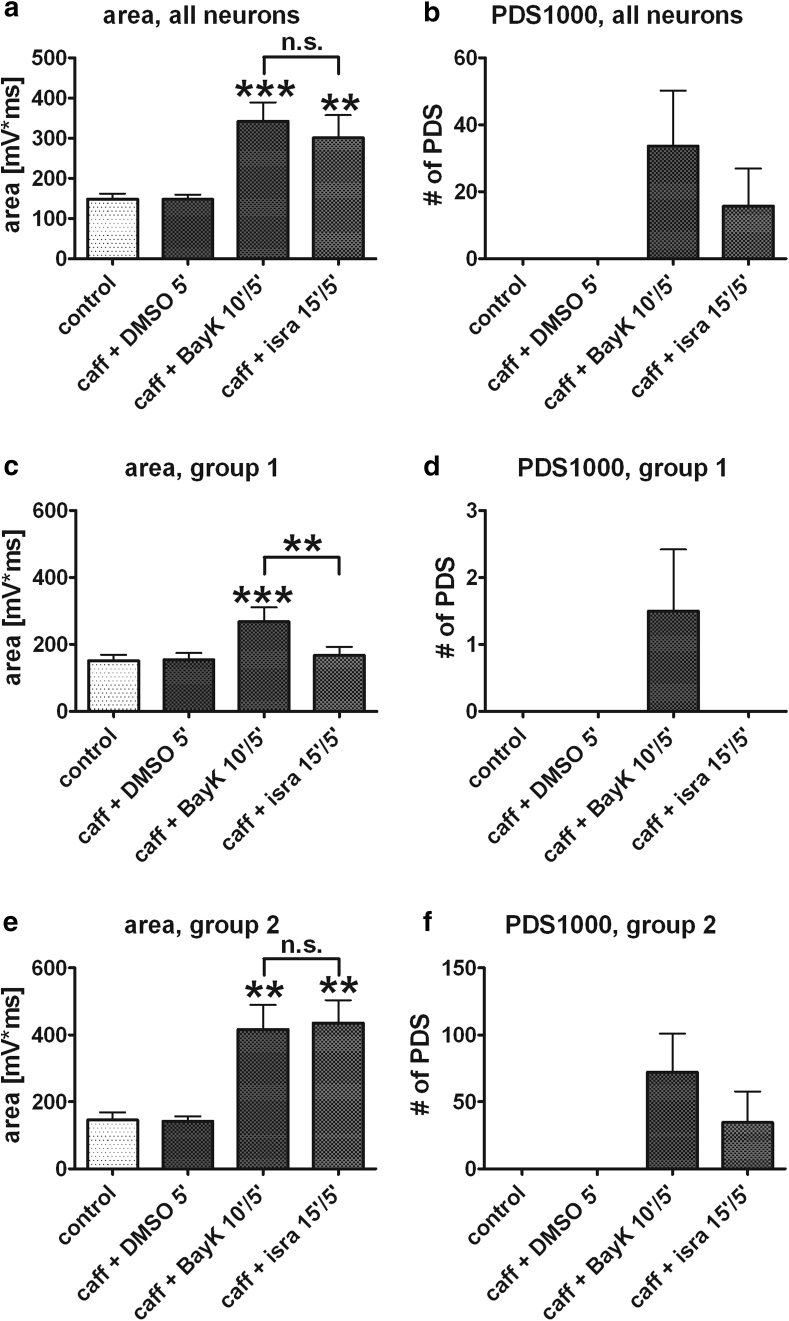
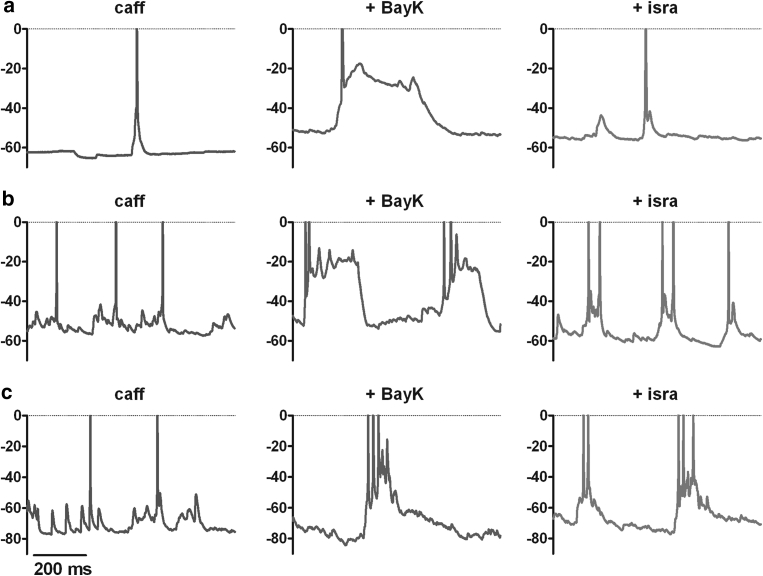
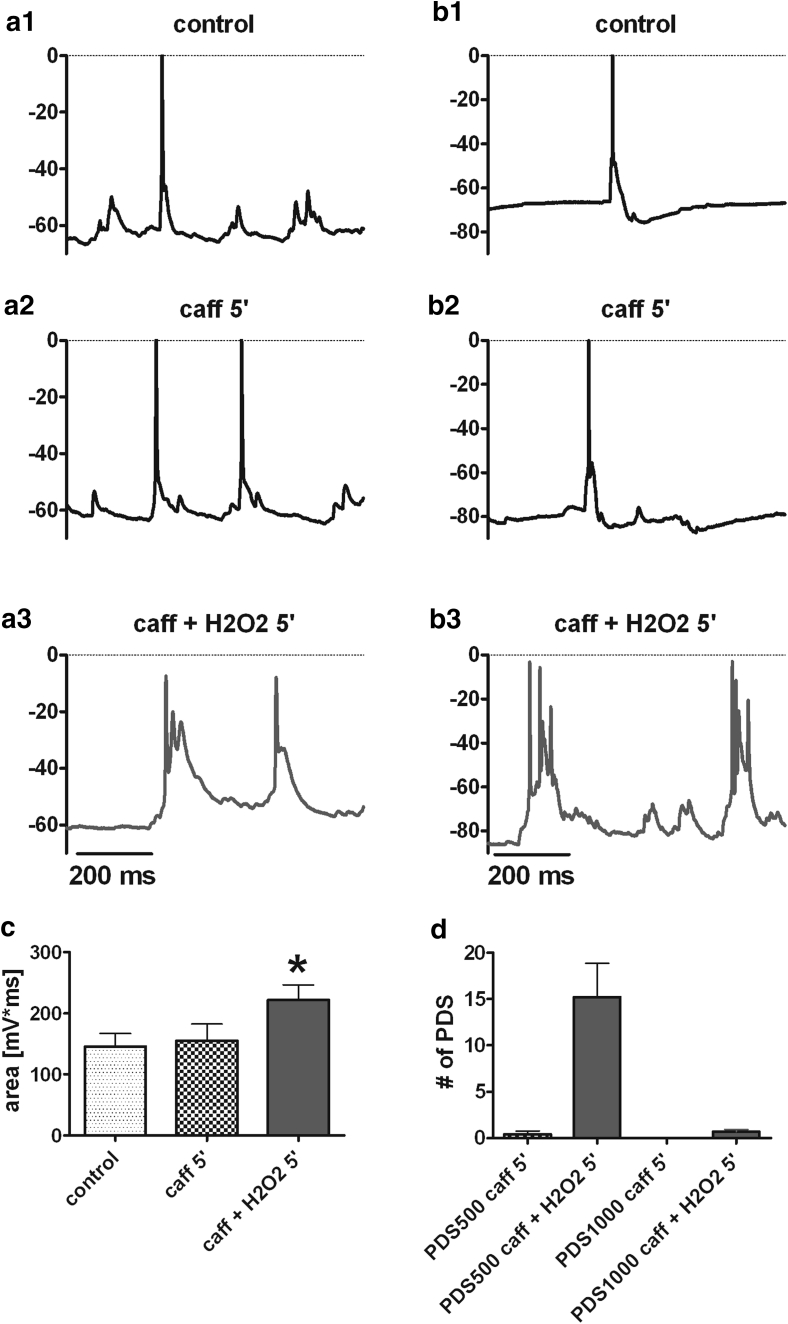
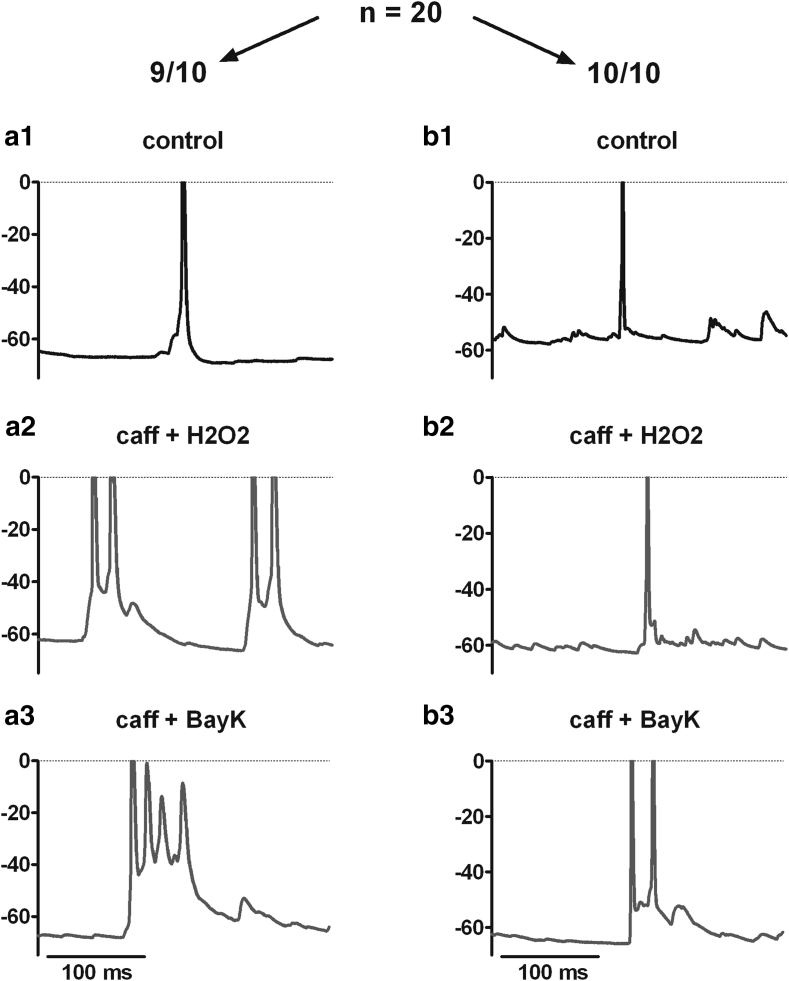
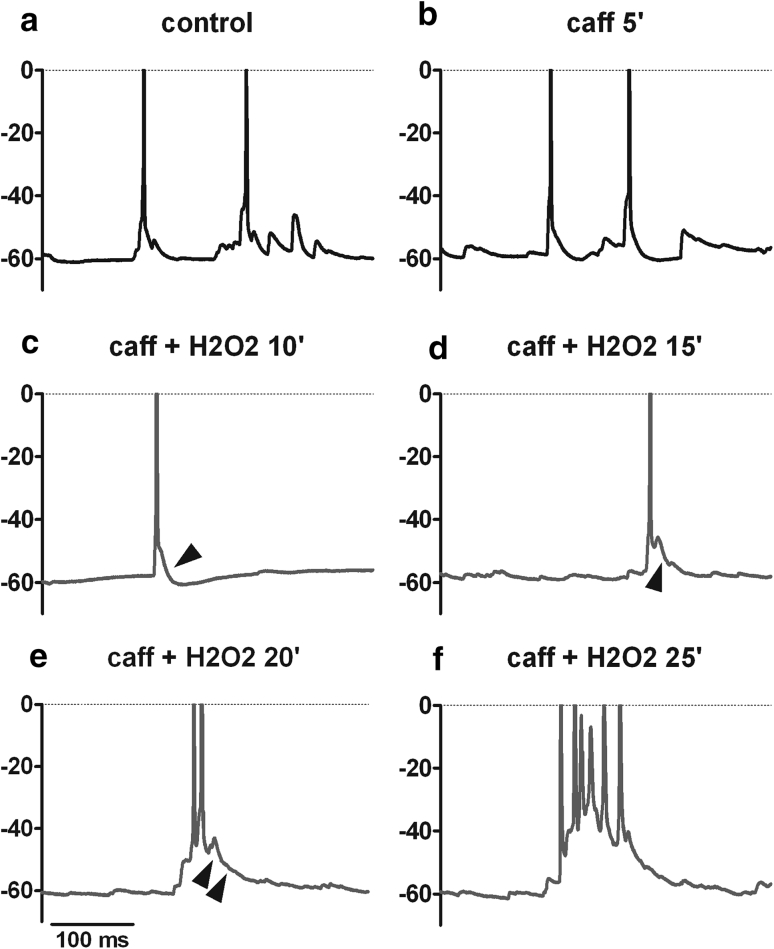
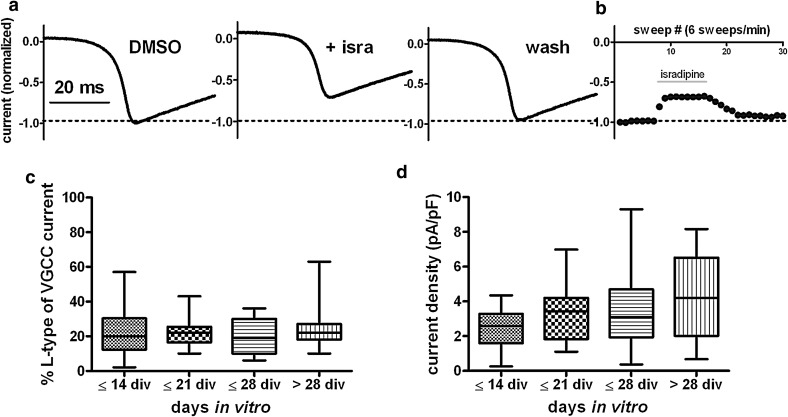
Neuronal L-type voltage-gated calcium channels (LTCCs) are involved in several physiological functions, but increased activity of LTCCs has been linked to pathology. Due to the coupling of LTCC-mediated Ca(2+) influx to Ca(2+)-dependent conductances, such as KCa or non-specific cation channels, LTCCs act as important regulators of neuronal excitability. Augmentation of after-hyperpolarizations may be one mechanism that shows how elevated LTCC activity can lead to neurological malfunctions. However, little is known about other impacts on electrical discharge activity. We used pharmacological up-regulation of LTCCs to address this issue on primary rat hippocampal neurons. Potentiation of LTCCs with Bay K8644 enhanced excitatory postsynaptic potentials to various degrees and eventually resulted in paroxysmal depolarization shifts (PDS). Under conditions of disturbed Ca(2+) homeostasis, PDS were evoked frequently upon LTCC potentiation. Exposing the neurons to oxidative stress using hydrogen peroxide also induced LTCC-dependent PDS. Hence, raising LTCC activity had unidirectional effects on brief electrical signals and increased the likeliness of epileptiform events. However, long-lasting seizure-like activity induced by various pharmacological means was affected by Bay K8644 in a bimodal manner, with increases in one group of neurons and decreases in another group. In each group, isradipine exerted the opposite effect. This suggests that therapeutic reduction in LTCC activity may have little beneficial or even adverse effects on long-lasting abnormal discharge activities. However, our data identify enhanced activity of LTCCs as one precipitating cause of PDS. Because evidence is continuously accumulating that PDS represent important elements in neuropathogenesis, LTCCs may provide valuable targets for neuroprophylactic therapy.
神经元 L 型电压门控钙通道(LTCCs)参与多种生理功能,但 LTCC 的活性增加与病理学有关。由于 LTCC 介导的 Ca2+内流与 Ca2+依赖性电导(如 KCa 或非特异性阳离子通道)偶联,LTCC 作为神经元兴奋性的重要调节剂。后超极化的增强可能是一种机制,表明升高的 LTCC 活性如何导致神经功能障碍。然而,对于其他对电放电活动的影响知之甚少。我们使用药理学上调 LTCC 来解决这个问题在原代大鼠海马神经元上。用 Bay K8644 增强 LTCC 可增强兴奋性突触后电位的程度不同,最终导致阵发性去极化转移(PDS)。在 Ca2+稳态失调的情况下,LTCC 增强后经常会诱发 PDS。使用过氧化氢使神经元暴露于氧化应激也会诱导 LTCC 依赖性 PDS。因此,提高 LTCC 活性对短暂的电信号有单向影响,并增加癫痫样事件的可能性。然而,各种药理学方法诱导的持续性癫痫样活动受 Bay K8644 以双模态方式影响,一组神经元增加,另一组神经元减少。在每组中,异搏定产生相反的效果。这表明 LTCC 活性的治疗性降低对持续性异常放电活动可能几乎没有有益甚至不利的影响。然而,我们的数据将 LTCCs 的活性增强确定为 PDS 的一个促成因素。由于不断有证据表明 PDS 是神经发病机制的重要因素,因此 LTCC 可能为神经保护治疗提供有价值的靶点。