Genetic Causes of Disease Group, Centre for Genomic Regulation (CRG), Barcelona, Spain.
PLoS One. 2013 May 21;8(5):e63377. doi: 10.1371/journal.pone.0063377. Print 2013.
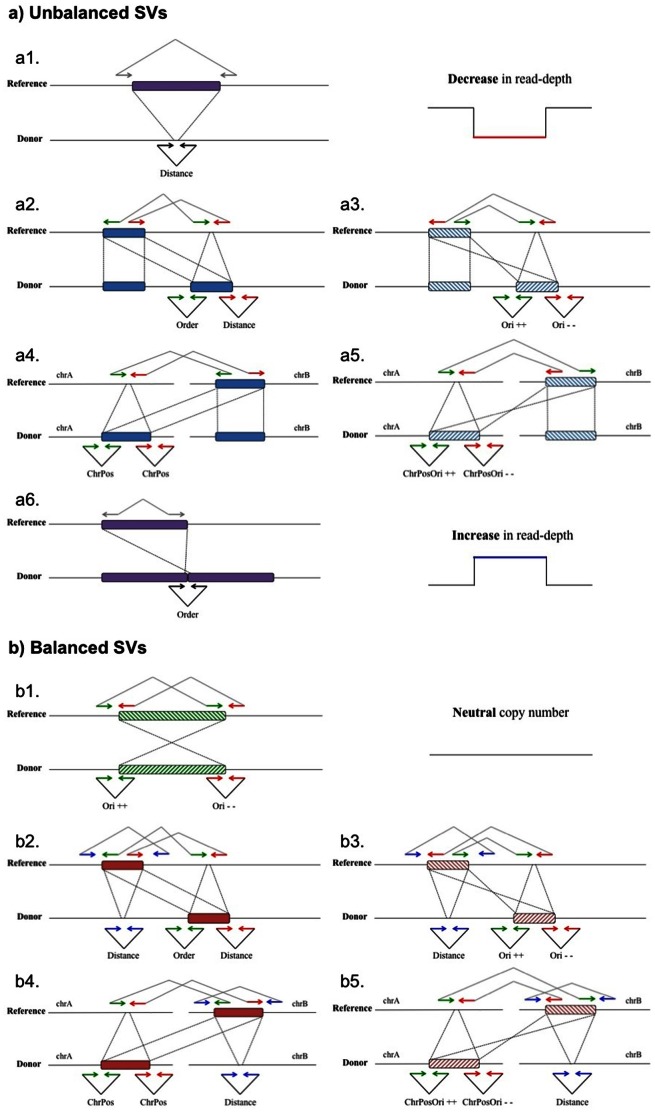
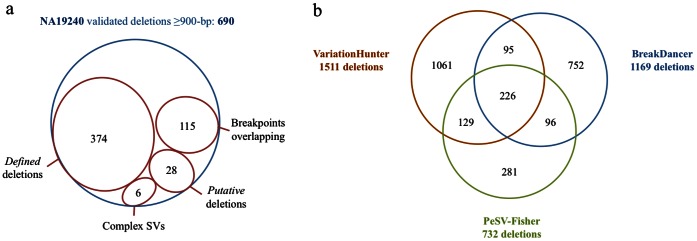
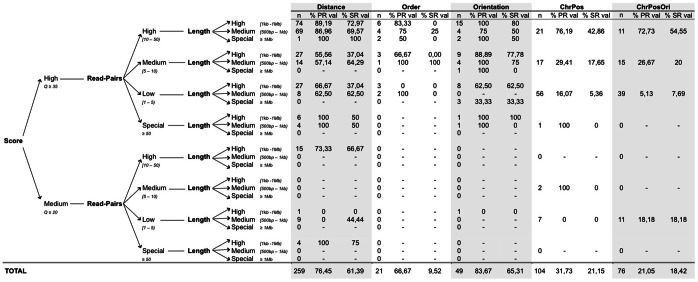
Next-generation sequencing technologies expedited research to develop efficient computational tools for the identification of structural variants (SVs) and their use to study human diseases. As deeper data is obtained, the existence of higher complexity SVs in some genomes becomes more evident, but the detection and definition of most of these complex rearrangements is still in its infancy. The full characterization of SVs is a key aspect for discovering their biological implications. Here we present a pipeline (PeSV-Fisher) for the detection of deletions, gains, intra- and inter-chromosomal translocations, and inversions, at very reasonable computational costs. We further provide comprehensive information on co-localization of SVs in the genome, a crucial aspect for studying their biological consequences. The algorithm uses a combination of methods based on paired-reads and read-depth strategies. PeSV-Fisher has been designed with the aim to facilitate identification of somatic variation, and, as such, it is capable of analysing two or more samples simultaneously, producing a list of non-shared variants between samples. We tested PeSV-Fisher on available sequencing data, and compared its behaviour to that of frequently deployed tools (BreakDancer and VariationHunter). We have also tested this algorithm on our own sequencing data, obtained from a tumour and a normal blood sample of a patient with chronic lymphocytic leukaemia, on which we have also validated the results by targeted re-sequencing of different kinds of predictions. This allowed us to determine confidence parameters that influence the reliability of breakpoint predictions.
PeSV-Fisher is available at http://gd.crg.eu/tools.
下一代测序技术加速了研究,以开发用于识别结构变体 (SVs) 的高效计算工具,并将其用于研究人类疾病。随着更深层次的数据的获得,一些基因组中更高复杂性 SVs 的存在变得更加明显,但这些复杂重排的大多数检测和定义仍处于起步阶段。SVs 的全面特征是发现其生物学意义的关键方面。在这里,我们提出了一种用于检测缺失、增益、染色体内和染色体间易位以及倒位的管道 (PeSV-Fisher),其计算成本非常合理。我们进一步提供了有关 SVs 在基因组中位置的综合信息,这是研究其生物学后果的关键方面。该算法使用了基于配对读取和读取深度策略的组合方法。PeSV-Fisher 的设计目的是方便识别体细胞变异,因此它能够同时分析两个或更多样本,并生成样本之间非共享变异的列表。我们在可用的测序数据上测试了 PeSV-Fisher,并将其行为与经常部署的工具 (BreakDancer 和 VariationHunter) 进行了比较。我们还在我们自己的测序数据上测试了该算法,该数据来自一名慢性淋巴细胞白血病患者的肿瘤和正常血液样本,我们还通过对不同类型的预测进行靶向重测序来验证了结果。这使我们能够确定影响断点预测可靠性的置信参数。
PeSV-Fisher 可在 http://gd.crg.eu/tools 上获得。