The first two authors contributed equally to the present study.
Hum Mol Genet. 2013 Oct 1;22(19):3869-82. doi: 10.1093/hmg/ddt242. Epub 2013 May 29.
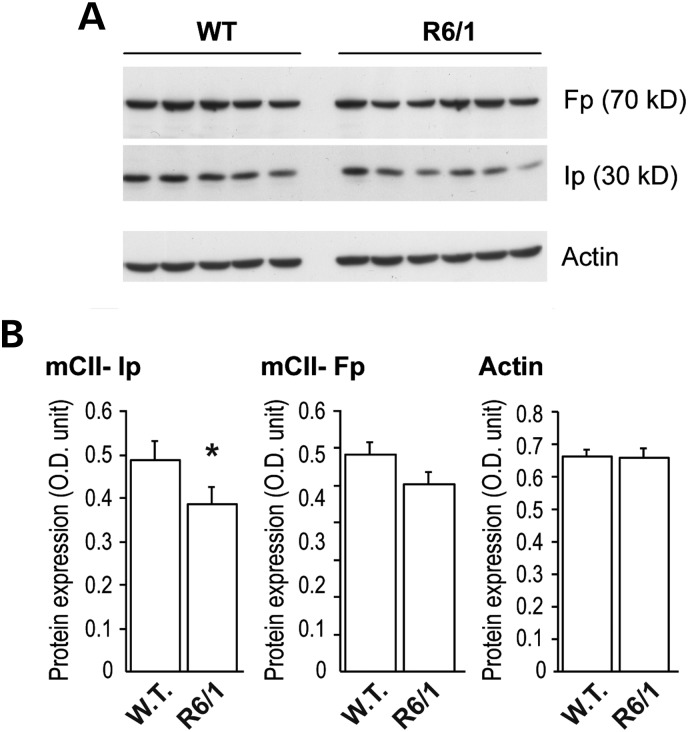
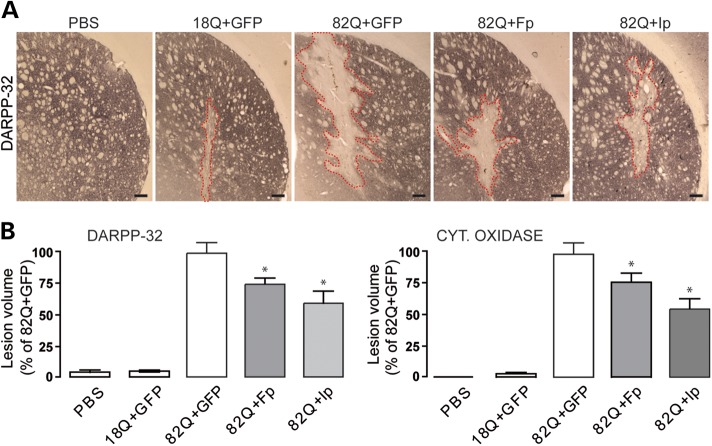
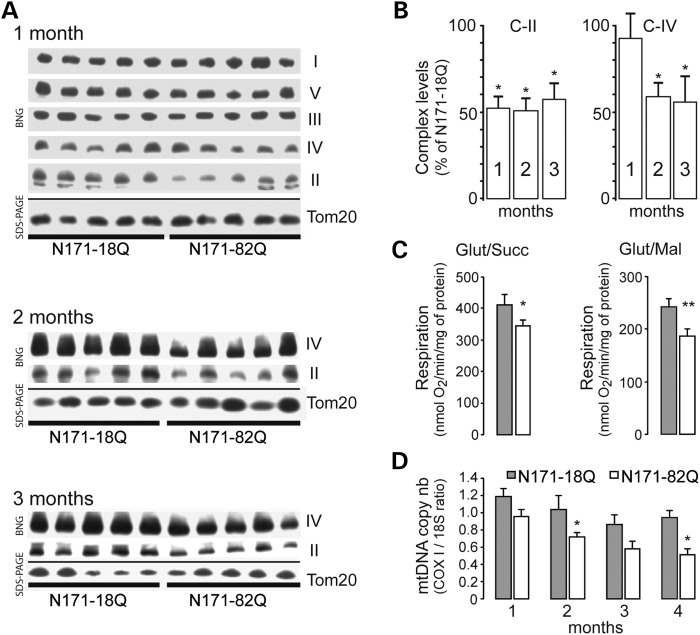
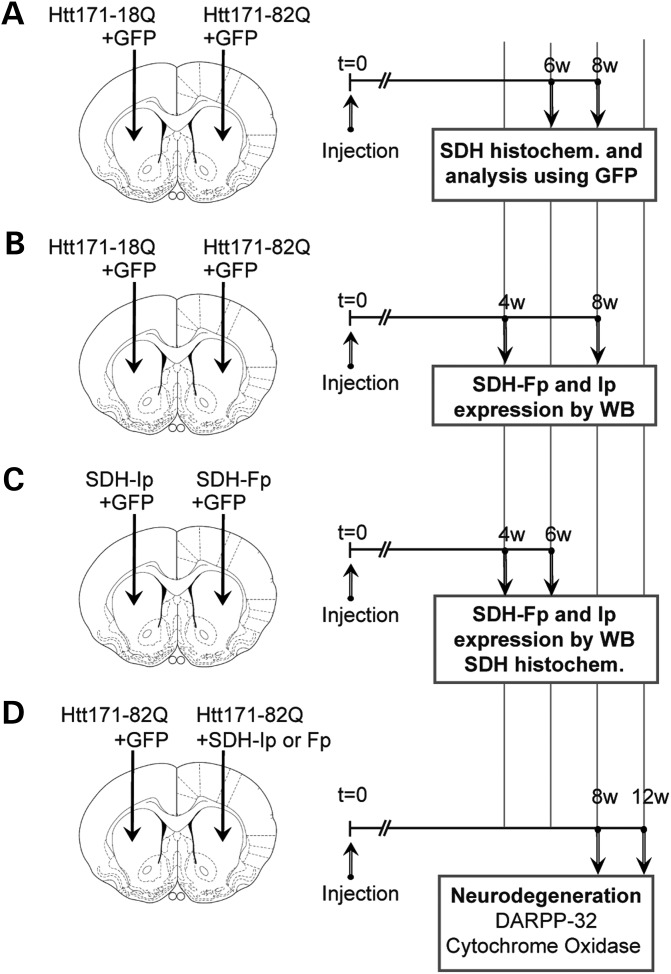
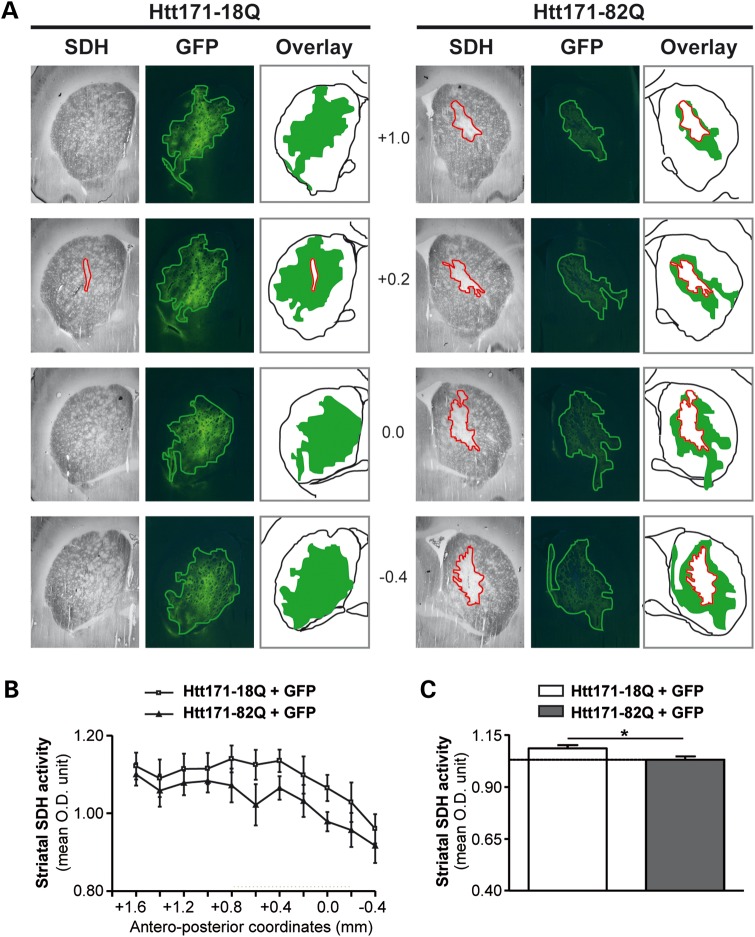
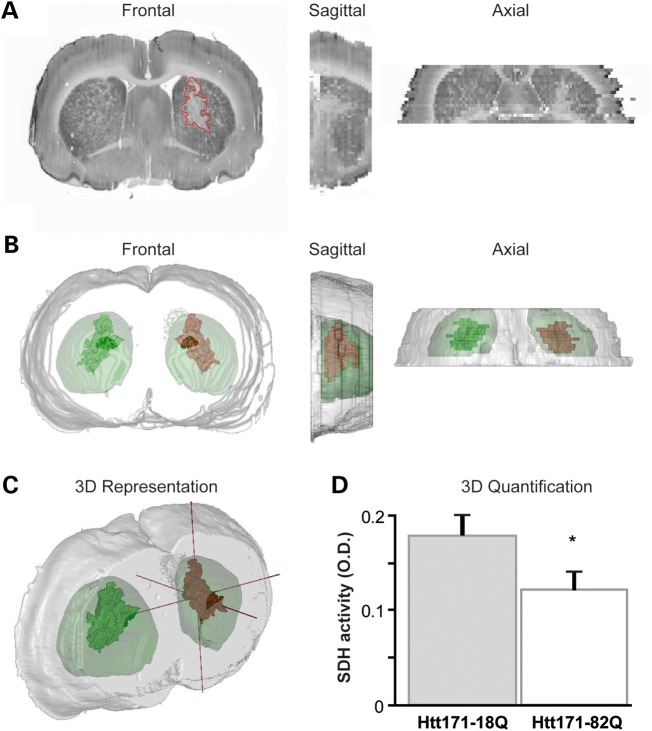
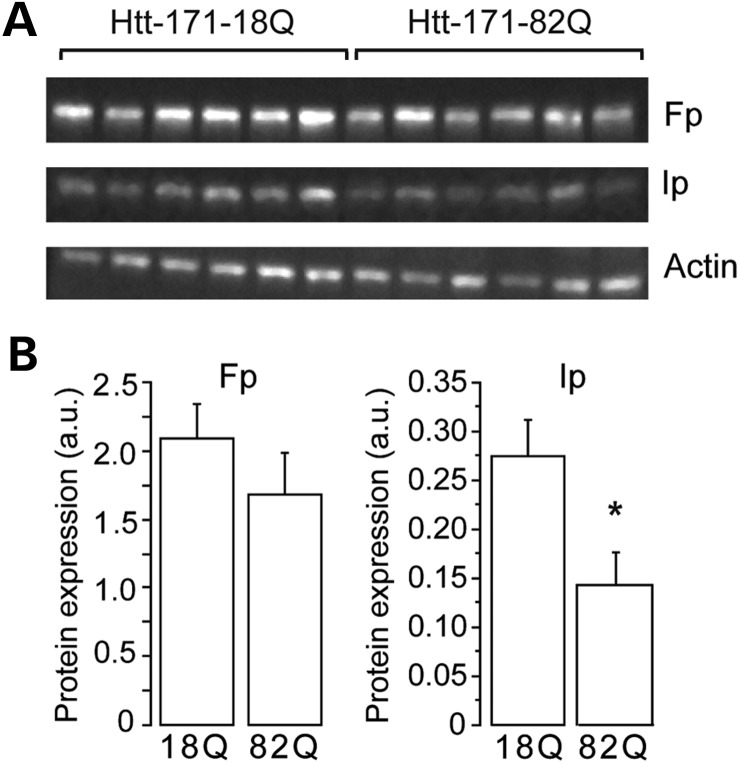
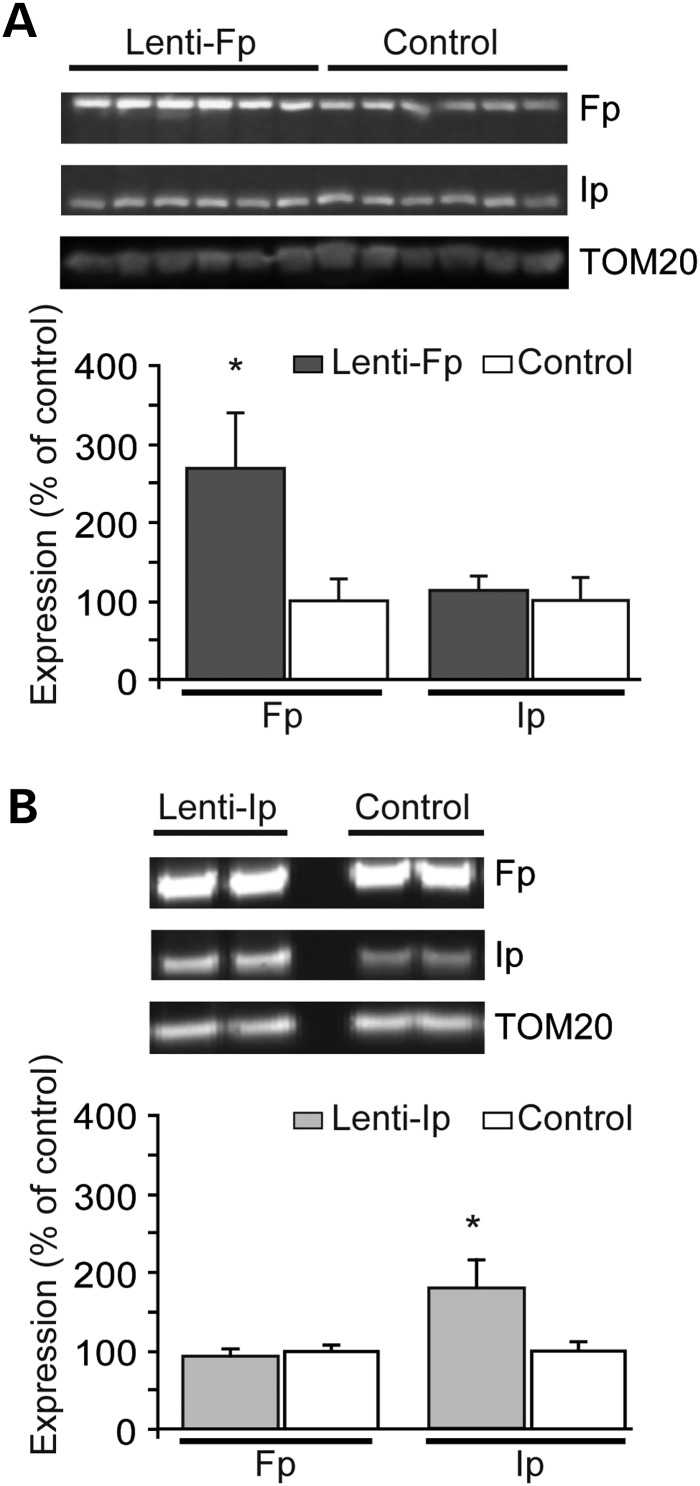
Huntington's disease (HD) is a neurodegenerative disorder caused by an abnormal expansion of a CAG repeat encoding a polyglutamine tract in the huntingtin (Htt) protein. The mutation leads to neuronal death through mechanisms which are still unknown. One hypothesis is that mitochondrial defects may play a key role. In support of this, the activity of mitochondrial complex II (C-II) is preferentially reduced in the striatum of HD patients. Here, we studied C-II expression in different genetic models of HD expressing N-terminal fragments of mutant Htt (mHtt). Western blot analysis showed that the expression of the 30 kDa Iron-Sulfur (Ip) subunit of C-II was significantly reduced in the striatum of the R6/1 transgenic mice, while the levels of the FAD containing catalytic 70 kDa subunit (Fp) were not significantly changed. Blue native gel analysis showed that the assembly of C-II in mitochondria was altered early in N171-82Q transgenic mice. Early loco-regional reduction in C-II activity and Ip protein expression was also demonstrated in a rat model of HD using intrastriatal injection of lentiviral vectors encoding mHtt. Infection of the rat striatum with a lentiviral vector coding the C-II Ip or Fp subunits induced a significant overexpression of these proteins that led to significant neuroprotection of striatal neurons against mHtt neurotoxicity. These results obtained in vivo support the hypothesis that structural and functional alterations of C-II induced by mHtt may play a critical role in the degeneration of striatal neurons in HD and that mitochondrial-targeted therapies may be useful in its treatment.
亨廷顿病(HD)是一种神经退行性疾病,由亨廷顿蛋白(Htt)中 CAG 重复序列异常扩展编码多聚谷氨酰胺链引起。突变通过未知的机制导致神经元死亡。一种假设是线粒体缺陷可能起关键作用。支持这一观点的是,HD 患者纹状体中线粒体复合物 II(C-II)的活性优先降低。在这里,我们研究了表达突变型 Htt(mHtt)N 端片段的不同 HD 遗传模型中的 C-II 表达。Western blot 分析表明,R6/1 转基因小鼠纹状体中 C-II 的 30 kDa 铁硫(Ip)亚基表达显著降低,而含有 FAD 的催化 70 kDa 亚基(Fp)的水平没有显著改变。蓝色天然凝胶分析表明,N171-82Q 转基因小鼠中线粒体中 C-II 的组装早期发生改变。使用编码 mHtt 的慢病毒载体在 HD 大鼠模型中也证明了 C-II 活性和 Ip 蛋白表达的早期局灶性降低。用编码 C-II Ip 或 Fp 亚基的慢病毒载体感染大鼠纹状体,可导致这些蛋白的显著过表达,从而对纹状体神经元对 mHtt 神经毒性的损伤产生显著的神经保护作用。这些体内获得的结果支持了这样一种假设,即 mHtt 诱导的 C-II 的结构和功能改变可能在 HD 纹状体神经元的退化中起关键作用,并且针对线粒体的治疗可能对其治疗有用。