School of Biomedical Sciences and Pharmacy, The University of Newcastle, Callaghan, New South Wales, Australia.
BMC Bioinformatics. 2013;14 Suppl 5(Suppl 5):S16. doi: 10.1186/1471-2105-14-S5-S16. Epub 2013 Apr 10.
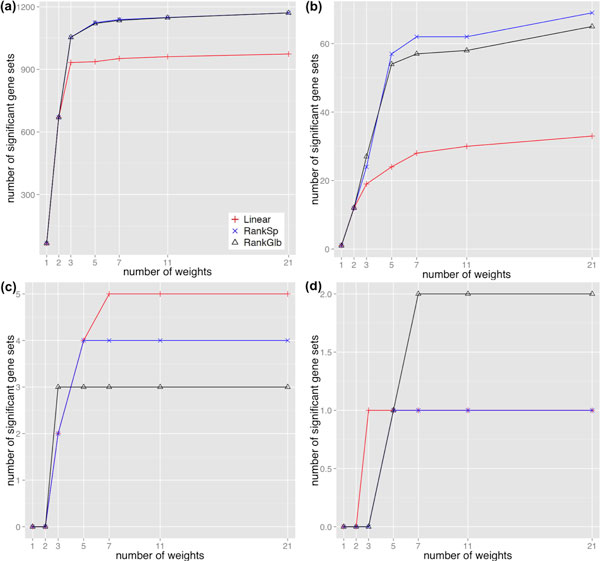
RNA-Seq has become a key technology in transcriptome studies because it can quantify overall expression levels and the degree of alternative splicing for each gene simultaneously. To interpret high-throughout transcriptome profiling data, functional enrichment analysis is critical. However, existing functional analysis methods can only account for differential expression, leaving differential splicing out altogether.
In this work, we present a novel approach to derive biological insight by integrating differential expression and splicing from RNA-Seq data with functional gene set analysis. This approach designated SeqGSEA, uses count data modelling with negative binomial distributions to first score differential expression and splicing in each gene, respectively, followed by two strategies to combine the two scores for integrated gene set enrichment analysis. Method comparison results and biological insight analysis on an artificial data set and three real RNA-Seq data sets indicate that our approach outperforms alternative analysis pipelines and can detect biological meaningful gene sets with high confidence, and that it has the ability to determine if transcription or splicing is their predominant regulatory mechanism.
By integrating differential expression and splicing, the proposed method SeqGSEA is particularly useful for efficiently translating RNA-Seq data to biological discoveries.
RNA-Seq 已成为转录组研究的关键技术,因为它可以同时定量每个基因的总体表达水平和可变剪接程度。为了解释高通量转录组分析数据,功能富集分析至关重要。然而,现有的功能分析方法只能考虑差异表达,完全忽略了差异剪接。
在这项工作中,我们提出了一种新方法,通过将 RNA-Seq 数据中的差异表达和剪接与功能基因集分析相结合,从转录组数据中得出生物学见解。这种方法命名为 SeqGSEA,它使用带有负二项分布的计数数据建模,首先分别对每个基因的差异表达和剪接进行评分,然后采用两种策略将这两个分数结合起来进行综合基因集富集分析。在人工数据集和三个真实 RNA-Seq 数据集上的方法比较结果和生物学见解分析表明,我们的方法优于替代分析管道,可以高置信度地检测具有生物学意义的基因集,并且能够确定转录或剪接是否是其主要的调控机制。
通过整合差异表达和剪接,所提出的方法 SeqGSEA 特别有助于将 RNA-Seq 数据高效转化为生物学发现。