Department of Electrical and Computer Engineering and Department of Computer Science and Engineering, University of California-San Diego, CA 92093, USA.
Bioinformatics. 2013 Aug 15;29(16):1953-62. doi: 10.1093/bioinformatics/btt338. Epub 2013 Jun 12.
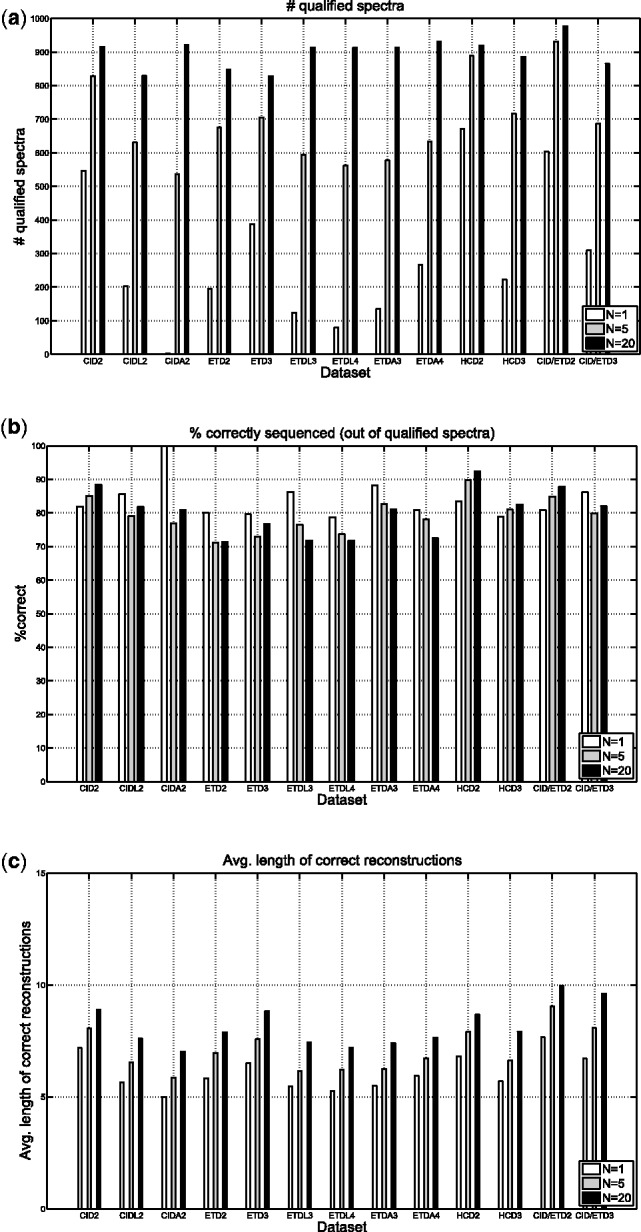
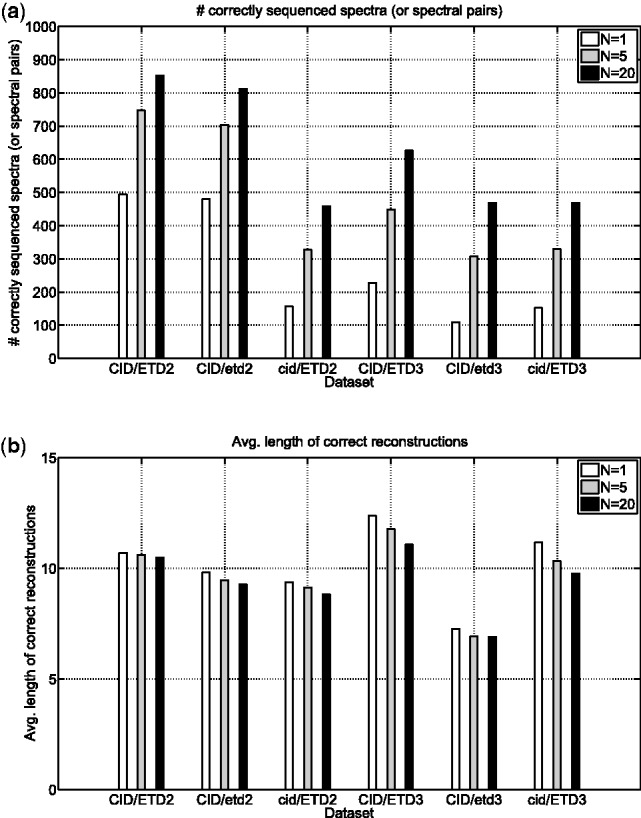
Mass spectrometry (MS) instruments and experimental protocols are rapidly advancing, but de novo peptide sequencing algorithms to analyze tandem mass (MS/MS) spectra are lagging behind. Although existing de novo sequencing tools perform well on certain types of spectra [e.g. Collision Induced Dissociation (CID) spectra of tryptic peptides], their performance often deteriorates on other types of spectra, such as Electron Transfer Dissociation (ETD), Higher-energy Collisional Dissociation (HCD) spectra or spectra of non-tryptic digests. Thus, rather than developing a new algorithm for each type of spectra, we develop a universal de novo sequencing algorithm called UniNovo that works well for all types of spectra or even for spectral pairs (e.g. CID/ETD spectral pairs). UniNovo uses an improved scoring function that captures the dependences between different ion types, where such dependencies are learned automatically using a modified offset frequency function.
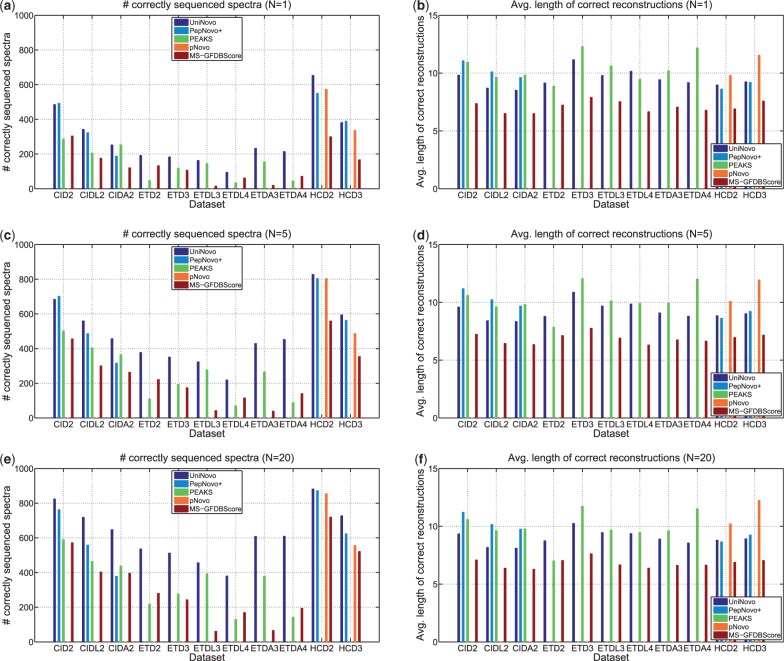
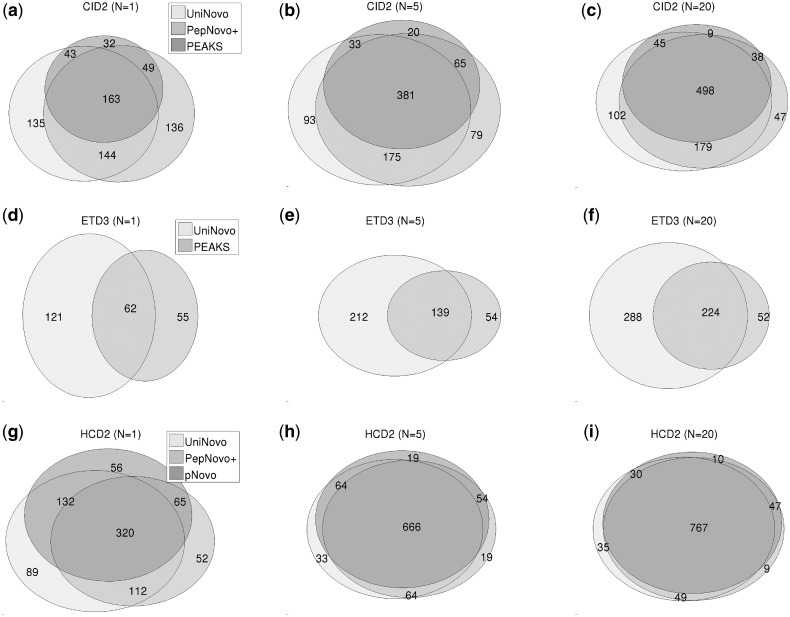
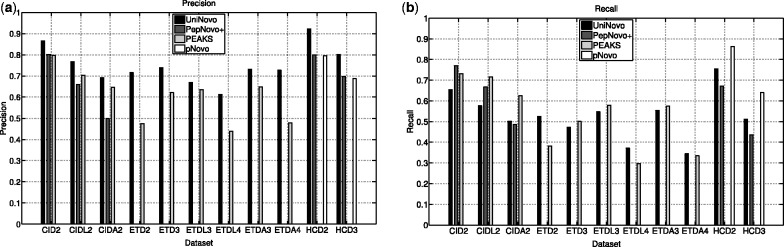
The performance of UniNovo is compared with PepNovo+, PEAKS and pNovo using various types of spectra. The results show that the performance of UniNovo is superior to other tools for ETD spectra and superior or comparable with others for CID and HCD spectra. UniNovo also estimates the probability that each reported reconstruction is correct, using simple statistics that are readily obtained from a small training dataset. We demonstrate that the estimation is accurate for all tested types of spectra (including CID, HCD, ETD, CID/ETD and HCD/ETD spectra of trypsin, LysC or AspN digested peptides).
UniNovo is implemented in JAVA and tested on Windows, Ubuntu and OS X machines. UniNovo is available at http://proteomics.ucsd.edu/Software/UniNovo.html along with the manual.
质谱(MS)仪器和实验方案正在迅速发展,但用于分析串联质谱(MS/MS)谱的从头多肽测序算法却滞后了。尽管现有的从头测序工具在某些类型的光谱[例如胰蛋白酶肽的碰撞诱导解离(CID)光谱]上表现良好,但它们在其他类型的光谱上的性能往往会恶化,例如电子转移解离(ETD)、更高能量的碰撞解离(HCD)光谱或非胰蛋白酶消化的光谱。因此,我们没有为每种类型的光谱开发新的算法,而是开发了一种通用的从头测序算法 UniNovo,它适用于所有类型的光谱,甚至适用于光谱对(例如 CID/ETD 光谱对)。UniNovo 使用改进的评分函数来捕获不同离子类型之间的依赖关系,其中这些依赖关系是使用修改后的偏移频率函数自动学习的。
使用各种类型的光谱比较了 UniNovo、PepNovo+、PEAKS 和 pNovo 的性能。结果表明,UniNovo 在 ETD 光谱上的性能优于其他工具,在 CID 和 HCD 光谱上的性能优于或可与其他工具相媲美。UniNovo 还使用简单的统计信息来估计每个报告重建的正确性的概率,这些信息可以从一个小的训练数据集轻松获得。我们证明,该估计对于所有测试类型的光谱(包括 CID、HCD、ETD、CID/ETD 和 HCD/ETD 胰蛋白酶、LysC 或 AspN 消化肽的光谱)都是准确的。
UniNovo 用 Java 实现,并在 Windows、Ubuntu 和 OS X 机器上进行了测试。UniNovo 可在 http://proteomics.ucsd.edu/Software/UniNovo.html 上获得,同时提供手册。