De Castro-Orós Isabel, Pocoví Miguel, Civeira Fernando
Lipid Unit and Laboratorio de Investigación Molecular, Hospital Universitario Miguel Servet, Instituto Aragonés de Ciencias de la Salud (I+CS), Zaragoza, Spain.
Appl Clin Genet. 2010 Aug 5;3:53-64. doi: 10.2147/tacg.s8285. Print 2010.
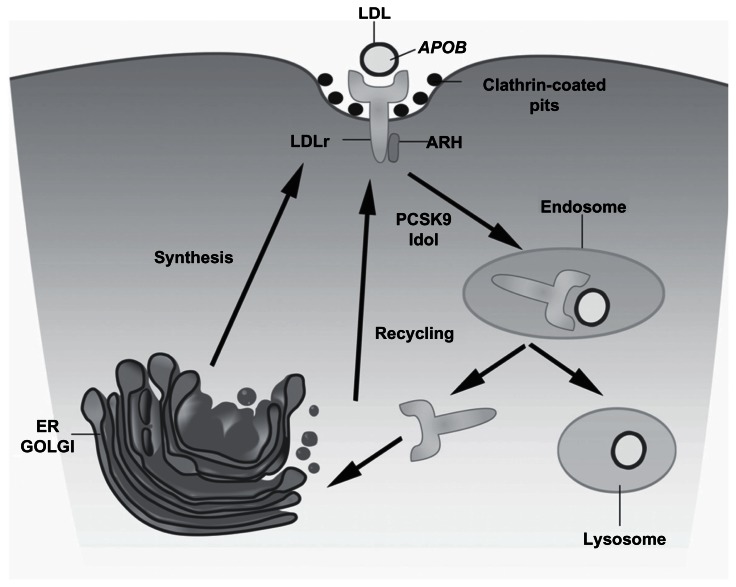
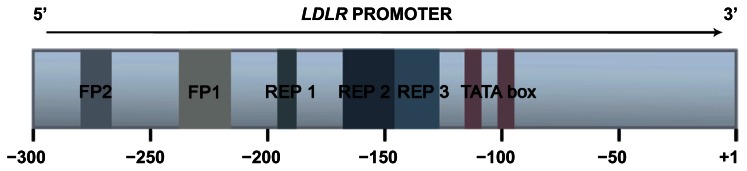
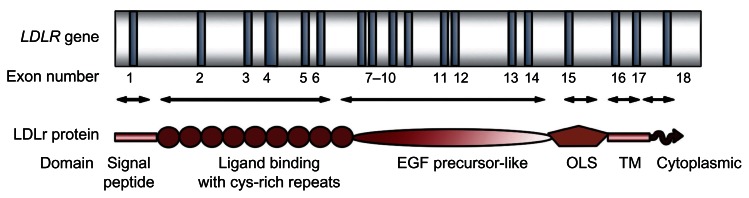
Familial hypercholesterolemia (FH) is a genetic disorder of lipoprotein metabolism characterized by high plasma concentrations of low-density lipoprotein cholesterol (LDLc), tendon xanthomas, and increased risk of premature coronary heart disease. FH is one of the most common inherited disorders; there are 10,000,000 people with FH worldwide, mainly heterozygotes. The most common FH cause is mutations along the entire gene that encode for LDL receptor (LDLR) protein, but it has been also described that mutations in apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 genes produce this phenotype. About 17%-33% of patients with a clinical diagnosis of monogenic hypercholesterolemia do not harbor any genetic cause in the known loci. Because FH has been considered as a public health problem, it is very important for an early diagnosis and treatment. Recent studies have demonstrated the influence of the LDLR mutation type in the FH phenotype, associating a more severe clinical phenotype and worse advanced carotid artherosclerosis in patients with null than those with receptor-defective mutations. Since 2004, a molecular FH diagnosis based on a genetic diagnostic platform (Lipochip(®); Progenika-Biopharma, Derio, Spain) has been developed. This analysis completes the adequate clinical diagnosis made by physicians. Our group has recently proposed new FH guidelines with the intention to facilitate the FH diagnosis. The treatment for this disease is based on the benefit of lowering LDLc and a healthy lifestyle. Actually, drug therapy is focused on using statins and combined therapy with ezetimibe and statins. This review highlights the recent progress made in genetics, diagnosis, and treatment for FH.
家族性高胆固醇血症(FH)是一种脂蛋白代谢的遗传性疾病,其特征为血浆低密度脂蛋白胆固醇(LDLc)浓度升高、肌腱黄色瘤以及早发冠心病风险增加。FH是最常见的遗传性疾病之一;全球有1000万人患有FH,主要为杂合子。FH最常见的病因是编码低密度脂蛋白受体(LDLR)蛋白的整个基因发生突变,但也有描述称载脂蛋白B(APOB)和枯草溶菌素/克新9型前蛋白转化酶基因的突变也会产生这种表型。临床诊断为单基因高胆固醇血症的患者中,约17%-33%在已知基因座未发现任何遗传病因。由于FH被视为一个公共卫生问题,早期诊断和治疗非常重要。最近的研究表明LDLR突变类型对FH表型有影响,与无功能突变患者相比,受体缺陷突变患者的临床表型更严重,晚期颈动脉粥样硬化更差。自2004年以来,基于基因诊断平台(Lipochip(®);西班牙德利奥的Progenika-Biopharma公司)开发了分子FH诊断方法。这种分析完善了医生做出的充分临床诊断。我们的团队最近提出了新的FH指南,旨在促进FH诊断。这种疾病的治疗基于降低LDLc的益处和健康的生活方式。实际上,药物治疗主要集中在使用他汀类药物以及依折麦布与他汀类药物的联合治疗。本综述重点介绍了FH在遗传学、诊断和治疗方面的最新进展。