Nephrologisches Zentrum, Medizinische Klinik und Poliklinik IV, Klinikum der Universität München, Ludwig-Maximilians-University Munich, Munich, Germany.
PLoS One. 2013 Jul 15;8(7):e68167. doi: 10.1371/journal.pone.0068167. Print 2013.
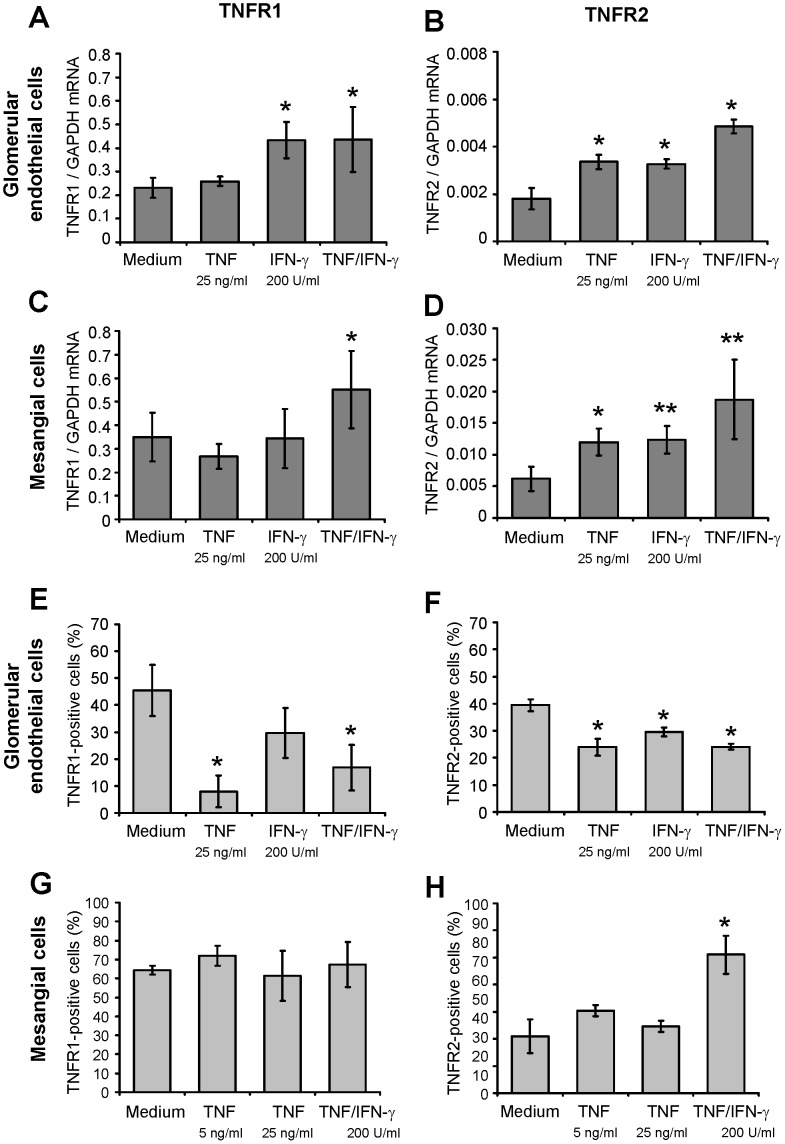
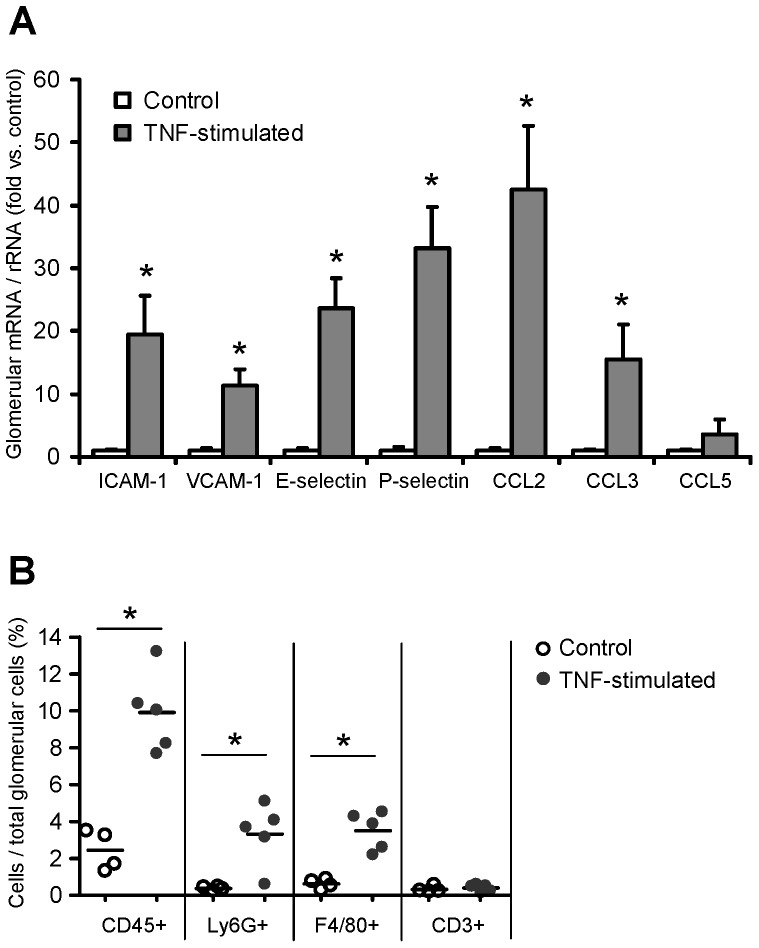
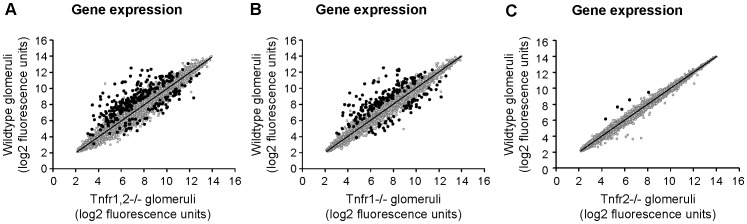
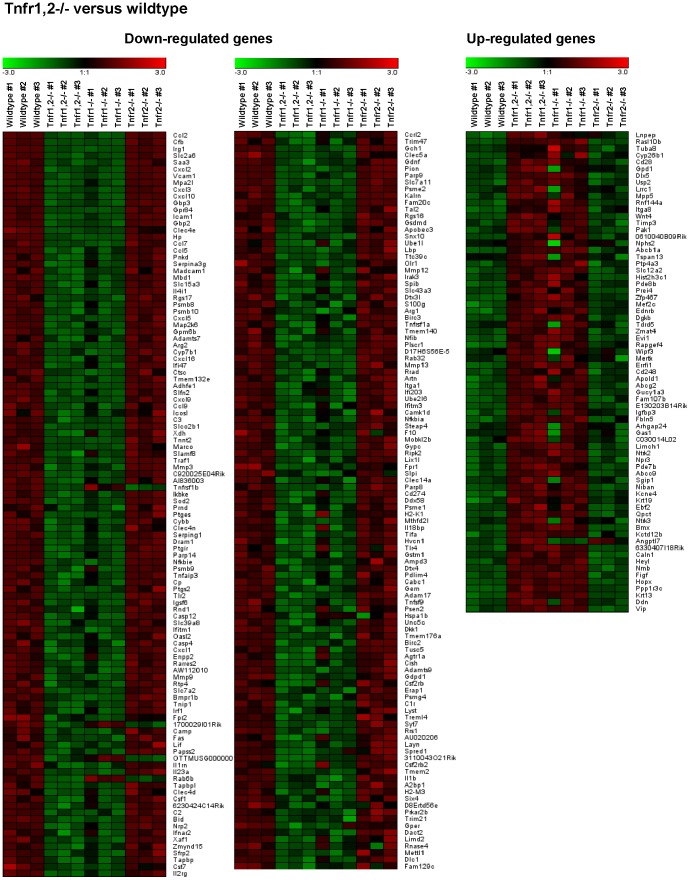
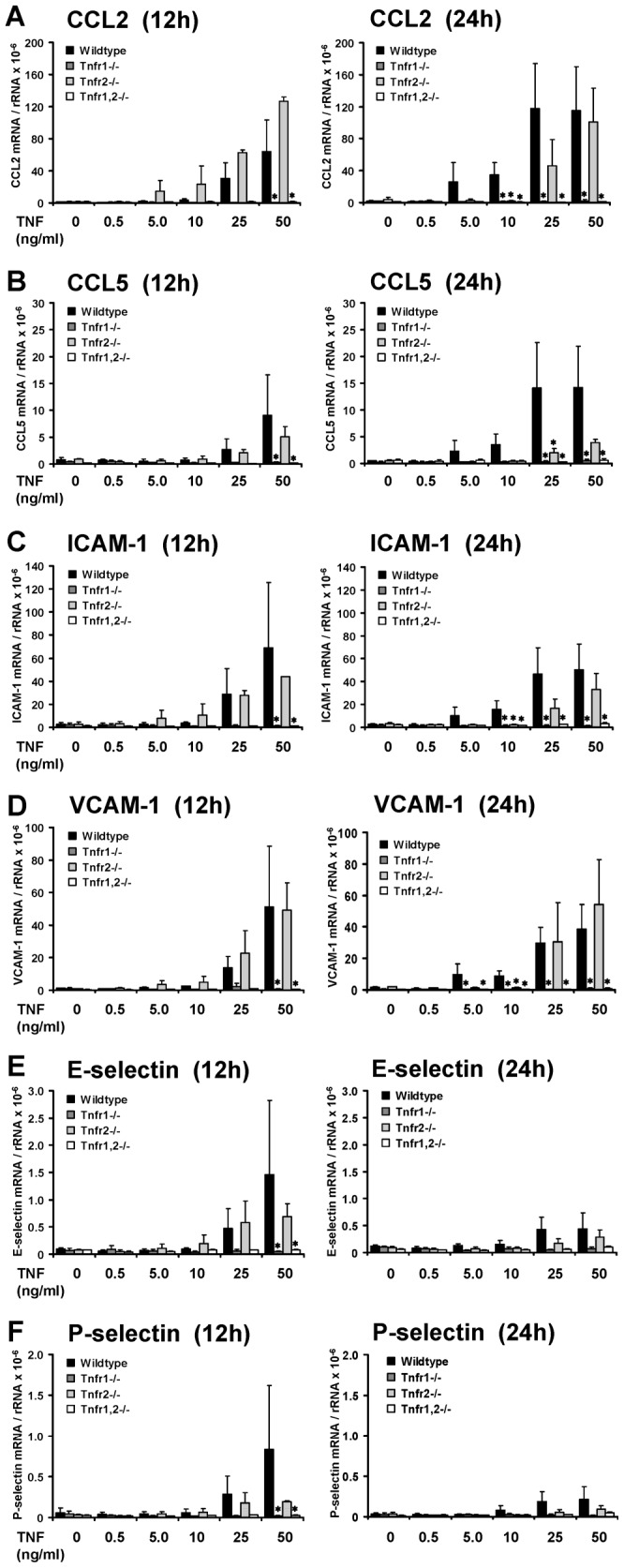
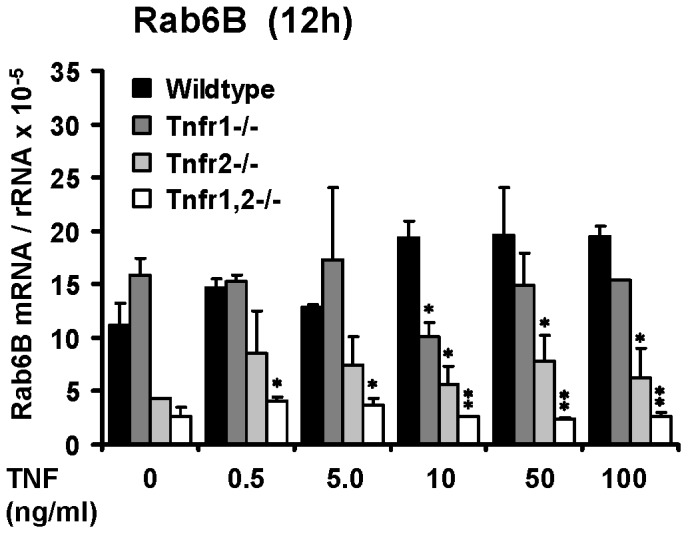
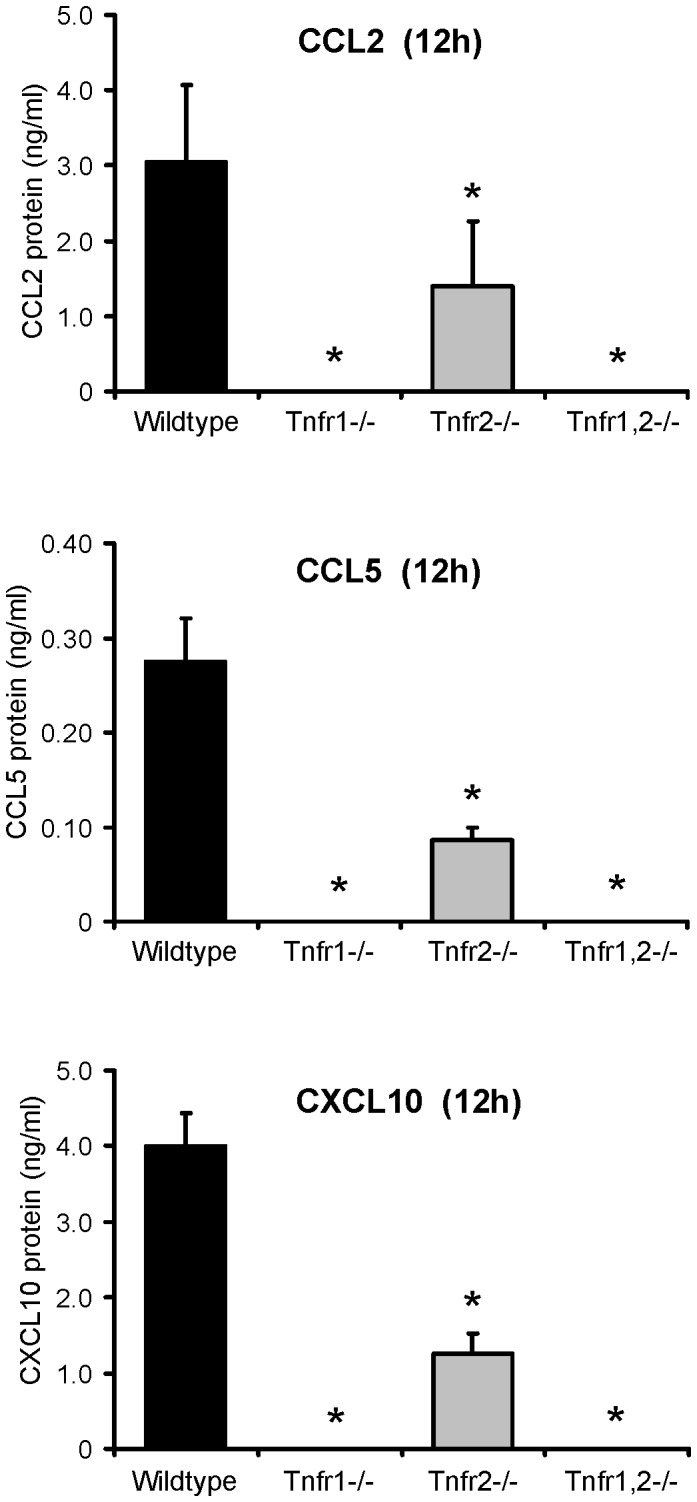
TNF is an important mediator of glomerulonephritis. The two TNF-receptors TNFR1 and TNFR2 contribute differently to glomerular inflammation in vivo, but specific mechanisms of TNFR-mediated inflammatory responses in glomeruli are unknown. We investigated their expression and function in murine kidneys, isolated glomeruli ex vivo, and glomerular cells in vitro. In normal kidney TNFR1 and TNFR2 were preferentially expressed in glomeruli. Expression of both TNFRs and TNF-induced upregulation of TNFR2 mRNA was confirmed in murine glomerular endothelial and mesangial cell lines. In vivo, TNF exposure rapidly induced glomerular accumulation of leukocytes. To examine TNFR-specific inflammatory responses in intrinsic glomerular cells but not infiltrating leukocytes we performed microarray gene expression profiling on intact glomeruli isolated from wildtype and Tnfr-deficient mice following exposure to soluble TNF ex vivo. Most TNF-induced effects were exclusively mediated by TNFR1, including induced glomerular expression of adhesion molecules, chemokines, complement factors and pro-apoptotic molecules. However, TNFR2 contributed to TNFR1-dependent mRNA expression of inflammatory mediators in glomeruli when exposed to low TNF concentrations. Chemokine secretion was absent in TNF-stimulated Tnfr1-deficient glomeruli, but also significantly decreased in glomeruli lacking TNFR2. In vivo, TNF-induced glomerular leukocyte infiltration was abrogated in Tnfr1-deficient mice, whereas Tnfr2-deficiency decreased mononuclear phagocytes infiltrates, but not neutrophils. These data demonstrate that activation of intrinsic glomerular cells by soluble TNF requires TNFR1, whereas TNFR2 is not essential, but augments TNFR1-dependent effects. Previously described TNFR2-dependent glomerular inflammation may therefore require TNFR2 activation by membrane-bound, but not soluble TNF.
肿瘤坏死因子(TNF)是肾小球肾炎的重要介质。两种 TNF 受体 TNFR1 和 TNFR2 在体内对肾小球炎症的贡献不同,但 TNFR 介导的肾小球炎症反应的特定机制尚不清楚。我们研究了它们在小鼠肾脏、离体肾小球和体外肾小球细胞中的表达和功能。在正常肾脏中,TNFR1 和 TNFR2 优先在肾小球中表达。在小鼠肾小球内皮细胞和系膜细胞系中,证实了两种 TNFR 的表达以及 TNF 诱导的 TNFR2 mRNA 上调。在体内,TNF 暴露迅速诱导白细胞在肾小球中积聚。为了研究内在肾小球细胞而不是浸润的白细胞中的 TNFR 特异性炎症反应,我们在暴露于可溶性 TNF 后,对从野生型和 Tnfr 缺陷型小鼠分离的完整肾小球进行了微阵列基因表达谱分析。大多数 TNF 诱导的作用仅由 TNFR1 介导,包括诱导肾小球表达粘附分子、趋化因子、补体因子和促凋亡分子。然而,当暴露于低 TNF 浓度时,TNFR2 有助于 TNFR1 依赖性炎症介质在肾小球中的表达。在 TNF 刺激的 Tnfr1 缺陷型肾小球中,趋化因子分泌缺失,但在缺乏 TNFR2 的肾小球中也显著减少。在体内,TNF 诱导的肾小球白细胞浸润在 Tnfr1 缺陷型小鼠中被阻断,而 Tnfr2 缺陷型小鼠减少单核吞噬细胞浸润,但不减少中性粒细胞浸润。这些数据表明,可溶性 TNF 激活内在肾小球细胞需要 TNFR1,而 TNFR2 不是必需的,但增强了 TNFR1 依赖性效应。因此,先前描述的 TNFR2 依赖性肾小球炎症可能需要膜结合而非可溶性 TNF 激活 TNFR2。