Balko Justin M, Stricker Thomas P, Arteaga Carlos L
Breast Cancer Res. 2013;15(4):209. doi: 10.1186/bcr3435.
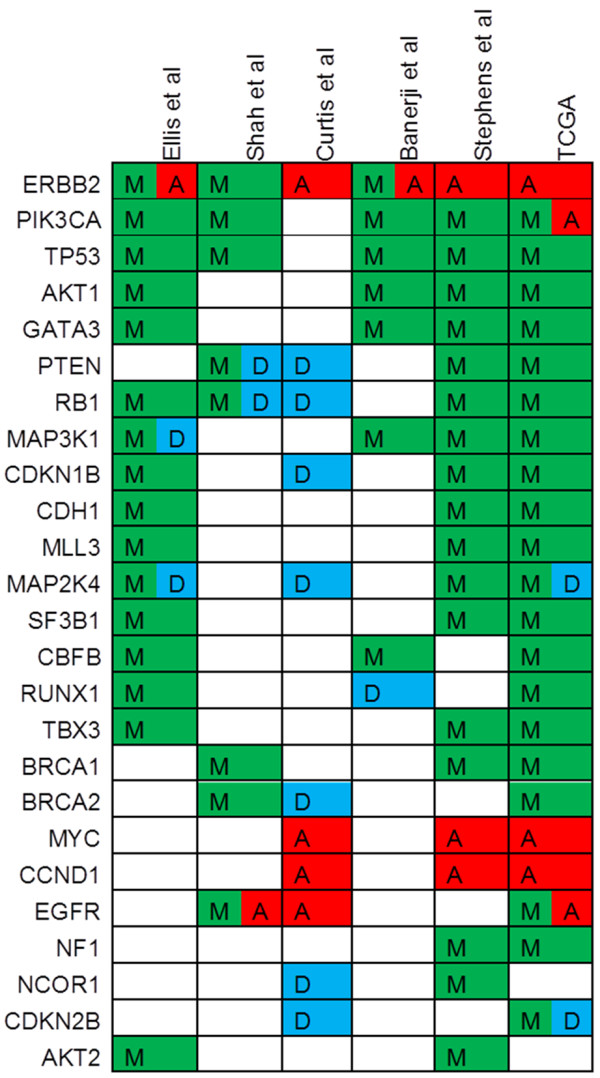
Recent advances in whole-genome technologies have supplied the field of cancer research with an overwhelming amount of molecular data. Improvements in massively parallel sequencing approaches have led to logarithmic decreases in costs, and so these methods are becoming almost commonplace in the analysis of clinical trials and other cohorts of interest. Furthermore, whole-transcriptome quantification by RNA sequencing is quickly replacing microarrays. However, older chip-based methodologies such as comparative genomic hybridization and single-nucleotide polymorphism arrays have benefited from this technological explosion and are now so accessible that they can be employed in increasingly larger cohorts of patients. The study of breast cancer lends itself particularly well to these technologies. It is the most commonly diagnosed neoplasm in women, giving rise to nearly 230,000 new cases each year. Many patients are given a diagnosis of early-stage disease, for which surgery is the standard of care. These attributes result in excellent availability of tissues for whole-genome/transcriptome analysis. The Cancer Genome Atlas project has generated comprehensive catalogs of publically available genomic breast cancer data. In addition, other studies employing the power of genomic technologies in medium to large cohorts were recently published. These data are now publically available for the generation of novel hypotheses. However, these studies differed in the methods, patient cohorts, and analytical techniques employed and represent complementary snapshots of the molecular underpinnings of breast cancer. Here, we will discuss the convergences and divergences of these reports as well as the scientific and clinical implications of their findings.
全基因组技术的最新进展为癌症研究领域提供了海量的分子数据。大规模平行测序方法的改进使得成本呈对数下降,因此这些方法在临床试验及其他相关队列分析中几乎变得司空见惯。此外,通过RNA测序进行的全转录组定量分析正在迅速取代微阵列。然而,诸如比较基因组杂交和单核苷酸多态性阵列等基于芯片的较老方法也从这一技术爆炸中受益,如今其使用非常便捷,可应用于越来越多的患者队列。乳腺癌研究尤其适合这些技术。它是女性中最常被诊断出的肿瘤,每年新增病例近23万例。许多患者被诊断为早期疾病,手术是其标准治疗方法。这些特点使得用于全基因组/转录组分析的组织极易获取。癌症基因组图谱项目已生成了公开可用的乳腺癌基因组数据综合目录。此外,最近还发表了其他一些在中大型队列中运用基因组技术的研究。这些数据现在可供公众用于提出新的假设。然而,这些研究在所用方法、患者队列和分析技术方面存在差异,代表了乳腺癌分子基础的互补性快照。在此,我们将讨论这些报告的异同及其研究结果的科学和临床意义。