Department of Biology, University of Konstanz, Konstanz, Germany.
PLoS One. 2013 Jul 31;8(7):e70327. doi: 10.1371/journal.pone.0070327. Print 2013.
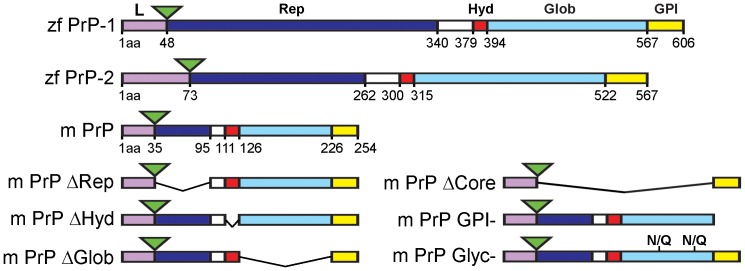
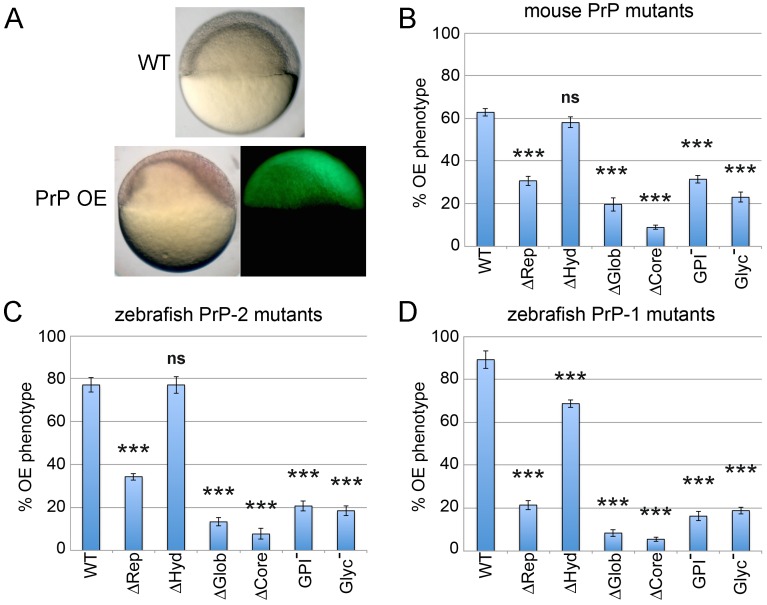
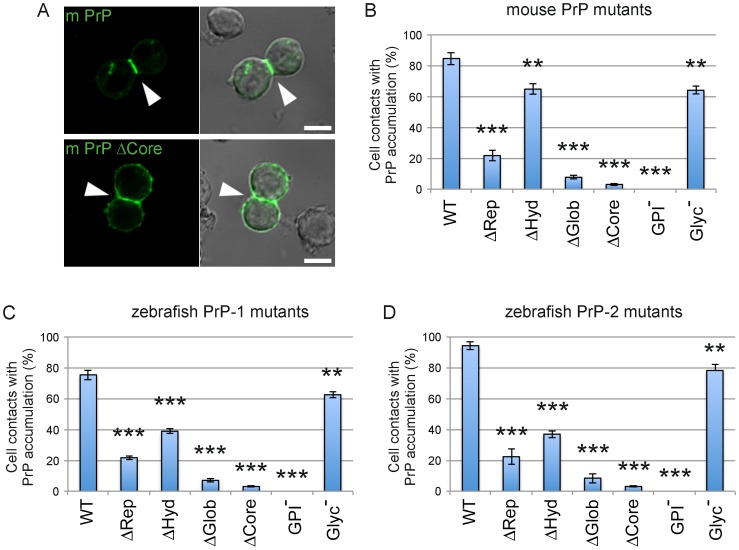
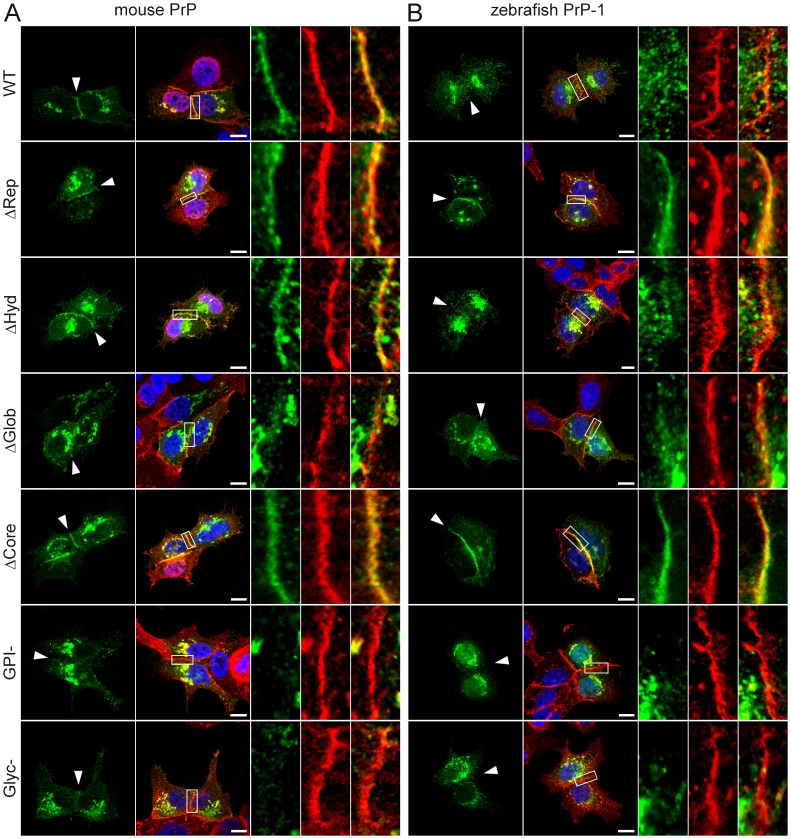
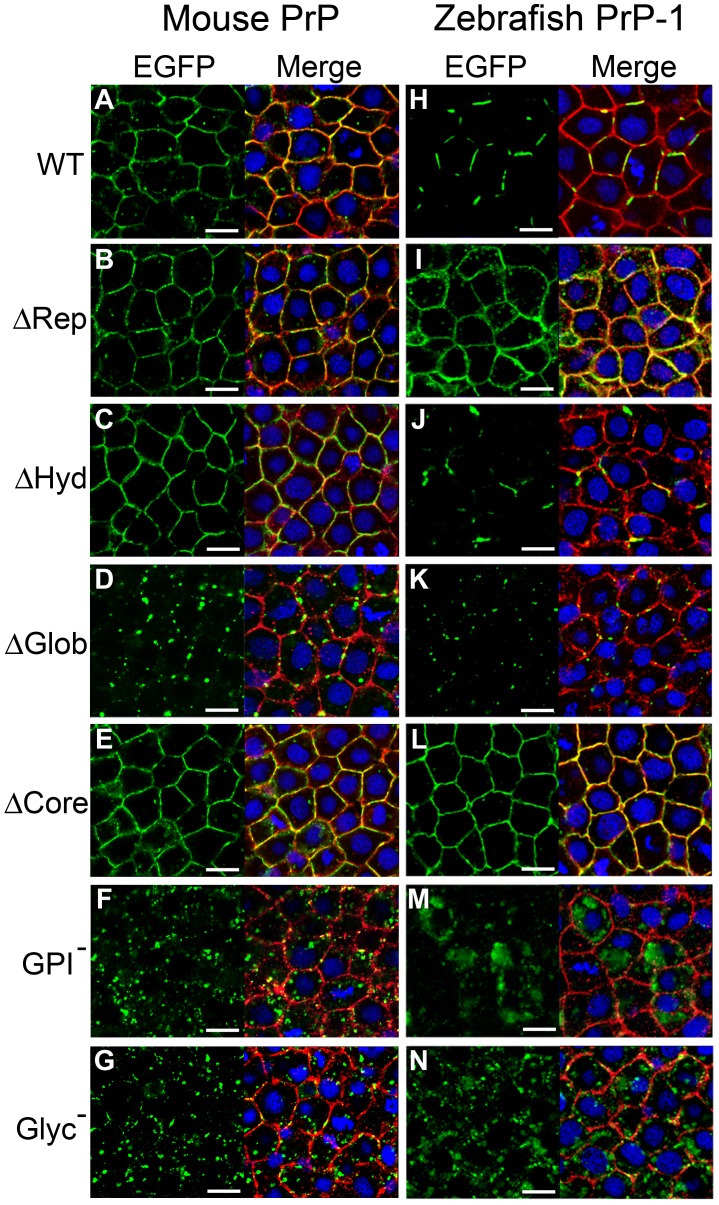
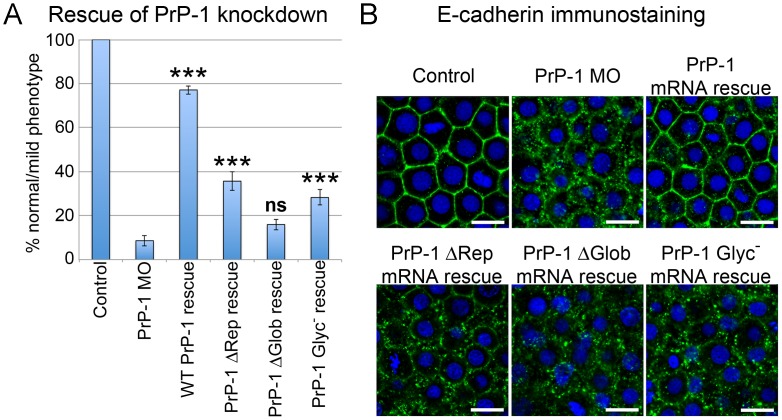
Analyses of cultured cells and transgenic mice expressing prion protein (PrP) deletion mutants have revealed that some properties of PrP -such as its ability to misfold, aggregate and trigger neurotoxicity- are controlled by discrete molecular determinants within its protein domains. Although the contributions of these determinants to PrP biosynthesis and turnover are relatively well characterized, it is still unclear how they modulate cellular functions of PrP. To address this question, we used two defined activities of PrP as functional readouts: 1) the recruitment of PrP to cell-cell contacts in Drosophila S2 and human MCF-7 epithelial cells, and 2) the induction of PrP embryonic loss- and gain-of-function phenotypes in zebrafish. Our results show that homologous mutations in mouse and zebrafish PrPs similarly affect their subcellular localization patterns as well as their in vitro and in vivo activities. Among PrP's essential features, the N-terminal leader peptide was sufficient to drive targeting of our constructs to cell contact sites, whereas lack of GPI-anchoring and N-glycosylation rendered them inactive by blocking their cell surface expression. Importantly, our data suggest that the ability of PrP to homophilically trans-interact and elicit intracellular signaling is primarily encoded in its globular domain, and modulated by its repetitive domain. Thus, while the latter induces the local accumulation of PrPs at discrete punctae along cell contacts, the former counteracts this effect by promoting the continuous distribution of PrP. In early zebrafish embryos, deletion of either domain significantly impaired PrP's ability to modulate E-cadherin cell adhesion. Altogether, these experiments relate structural features of PrP to its subcellular distribution and in vivo activity. Furthermore, they show that despite their large evolutionary history, the roles of PrP domains and posttranslational modifications are conserved between mouse and zebrafish.
对表达朊病毒蛋白 (PrP) 缺失突变体的培养细胞和转基因小鼠的分析表明,PrP 的某些性质——例如其错误折叠、聚集和引发神经毒性的能力——由其蛋白结构域内的离散分子决定因素控制。尽管这些决定因素对 PrP 生物合成和周转的贡献相对较好地描述,但仍不清楚它们如何调节 PrP 的细胞功能。为了解决这个问题,我们使用 PrP 的两个定义的活性作为功能读出:1)PrP 在果蝇 S2 和人 MCF-7 上皮细胞中与细胞-细胞接触的募集,以及 2)在斑马鱼中诱导 PrP 胚胎缺失和获得功能表型。我们的结果表明,小鼠和斑马鱼 PrP 中的同源突变类似地影响它们的亚细胞定位模式以及它们的体外和体内活性。在 PrP 的基本特征中,N 端前导肽足以驱动我们的构建体靶向细胞接触部位,而缺乏 GPI-锚定和 N-糖基化通过阻止其细胞表面表达使它们失活。重要的是,我们的数据表明,PrP 同型相互作用和引发细胞内信号的能力主要由其球形结构域编码,并由其重复结构域调节。因此,虽然后者导致 PrP 在细胞接触处的离散点状处局部积累,但前者通过促进 PrP 的连续分布来抵消这种效应。在早期的斑马鱼胚胎中,缺失任何一个结构域都会显著损害 PrP 调节 E-钙粘蛋白细胞黏附的能力。总之,这些实验将 PrP 的结构特征与其亚细胞分布和体内活性联系起来。此外,它们表明,尽管它们具有很大的进化历史,但 PrP 结构域和翻译后修饰的作用在小鼠和斑马鱼之间是保守的。