Evolutionary Genetics, Leibniz-Institute for Zoo and Wildlife Research, Alfred-Kowalke-Straße 17, D-10315 Berlin, Germany.
BMC Genomics. 2013 Aug 9;14:542. doi: 10.1186/1471-2164-14-542.
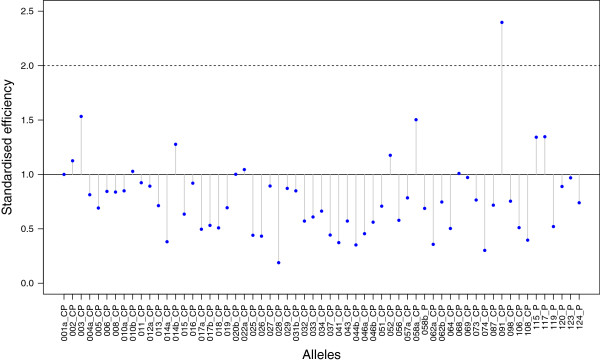
The Major Histocompatibility Complex (MHC) is the most important genetic marker to study patterns of adaptive genetic variation determining pathogen resistance and associated life history decisions. It is used in many different research fields ranging from human medical, molecular evolutionary to functional biodiversity studies. Correct assessment of the individual allelic diversity pattern and the underlying structural sequence variation is the basic requirement to address the functional importance of MHC variability. Next-generation sequencing (NGS) technologies are likely to replace traditional genotyping methods to a great extent in the near future but first empirical studies strongly indicate the need for a rigorous quality control pipeline. Strict approaches for data validation and allele calling to distinguish true alleles from artefacts are required.
We developed the analytical methodology and validated a data processing procedure which can be applied to any organism. It allows the separation of true alleles from artefacts and the evaluation of genotyping reliability, which in addition to artefacts considers for the first time the possibility of allelic dropout due to unbalanced amplification efficiencies across alleles. Finally, we developed a method to assess the confidence level per genotype a-posteriori, which helps to decide which alleles and individuals should be included in any further downstream analyses. The latter method could also be used for optimizing experiment designs in the future.
Combining our workflow with the study of amplification efficiency offers the chance for researchers to evaluate enormous amounts of NGS-generated data in great detail, improving confidence over the downstream analyses and subsequent applications.
主要组织相容性复合体 (MHC) 是研究决定病原体抗性和相关生活史决策的适应性遗传变异模式的最重要遗传标记。它被用于许多不同的研究领域,从人类医学、分子进化到功能生物多样性研究。正确评估个体等位基因多样性模式和潜在的结构序列变异是解决 MHC 变异性功能重要性的基本要求。下一代测序 (NGS) 技术很可能在不久的将来在很大程度上取代传统的基因分型方法,但首先进行的实证研究强烈表明需要严格的质量控制流程。需要严格的方法来验证数据和等位基因调用,以区分真实等位基因和伪影。
我们开发了一种分析方法,并验证了一种数据处理程序,该程序可应用于任何生物体。它允许从伪影中分离真实等位基因,并评估基因分型的可靠性,除了伪影之外,它还首次考虑了由于等位基因之间扩增效率不平衡而导致等位基因丢失的可能性。最后,我们开发了一种方法来评估每个基因型的置信度水平,这有助于决定应该在任何进一步的下游分析中包含哪些等位基因和个体。该方法也可以用于优化未来的实验设计。
将我们的工作流程与扩增效率的研究相结合,为研究人员提供了评估大量 NGS 生成数据的机会,从而提高了对下游分析和后续应用的信心。