Cancer Science Institute of Singapore, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
PLoS One. 2013 Aug 5;8(8):e70891. doi: 10.1371/journal.pone.0070891. Print 2013.
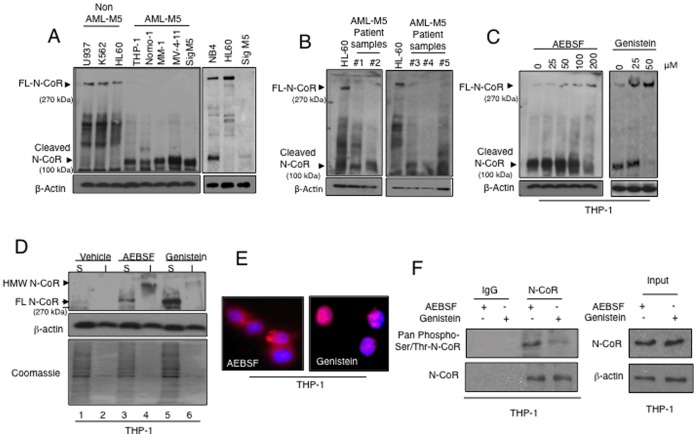
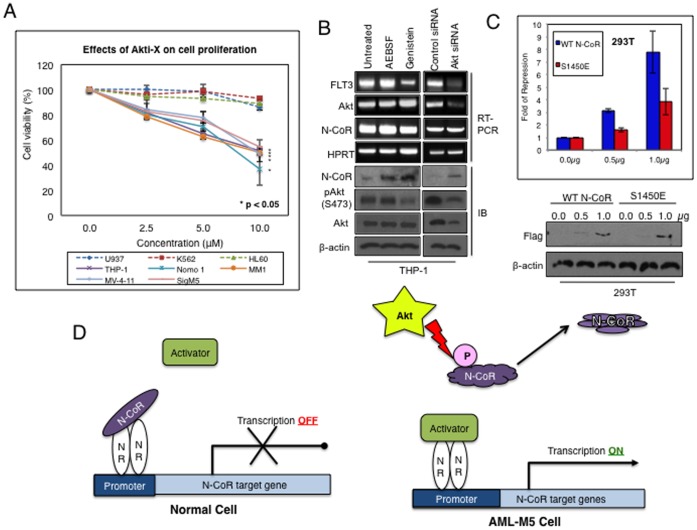
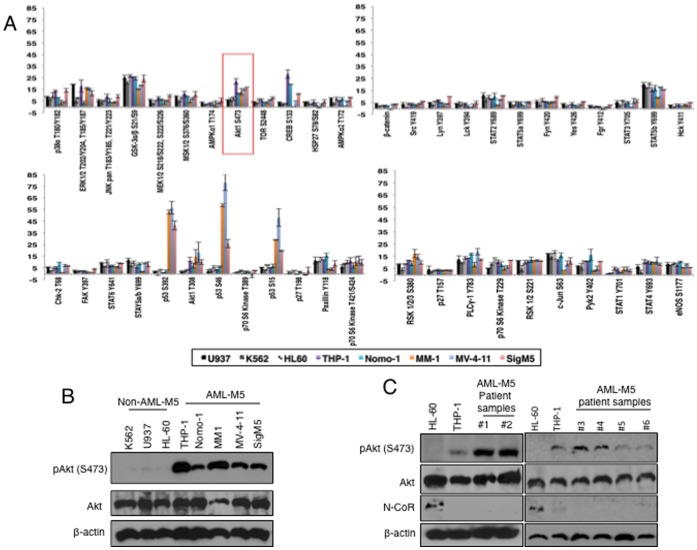
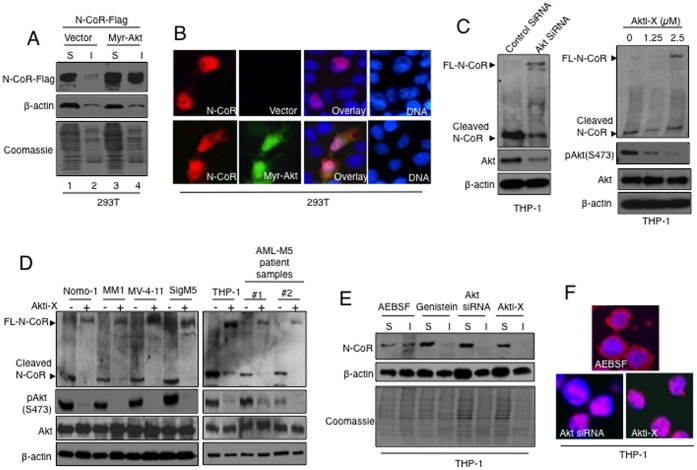
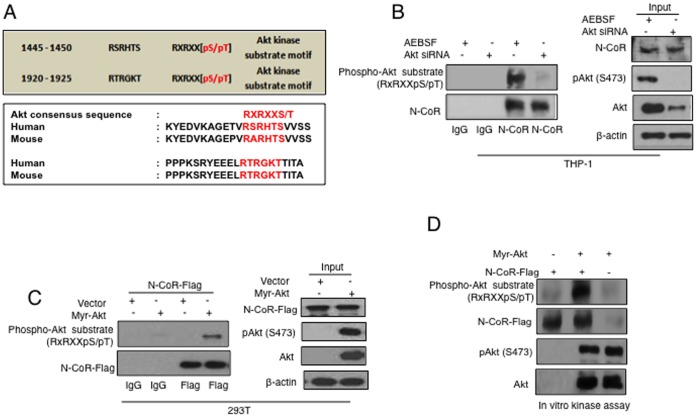
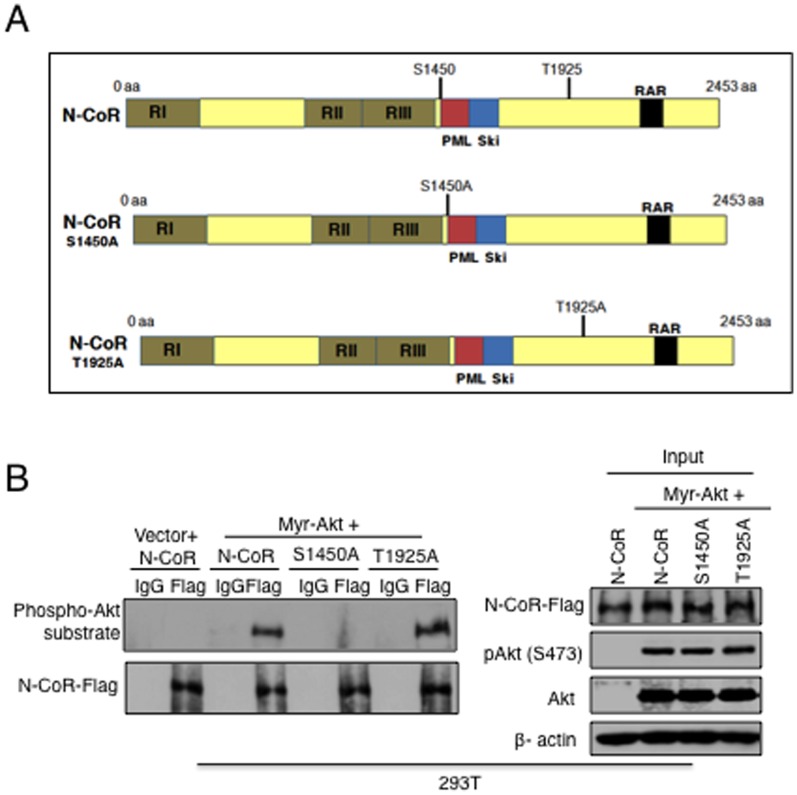
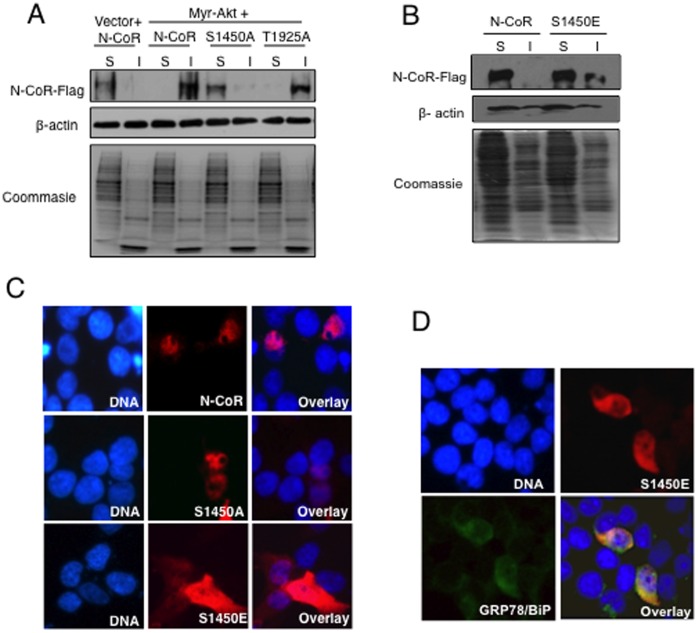
The nuclear receptor co-repressor (N-CoR) is a key component of the generic co-repressor complex that plays an important role in the control of cellular growth and differentiation. As shown by us recently, the growth suppressive function of N-CoR largely relies on its capacity to repress Flt3, a key regulator of cellular gorwth during normal and malignant hematopoesis. We further demonstrated how de-repression of Flt3 due to the misfolded conformation dependent loss (MCDL) of N-CoR contributed to malignant growth in acute myeloid leukemia (AML). However, the molecular mechanism underlying the MCDL of N-CoR and its implication in AML pathogenesis is not fully understood. Here, we report that Akt-induced phosphorylation of N-CoR at the consensus Akt motif is crucial for its misfolding and subsequent loss in AML (AML-M5). N-CoR displayed significantly higher level of serine specific phosphorylation in almost all AML-M5 derived cells and was subjected to processing by AML-M5 specific aberrant protease activity. To identify the kinase linked to N-CoR phosphorylation, a library of activated kinases was screened with the extracts of AML cells; leading to the identification of Akt as the putative kinase linked to N-CoR phosphorylation. Consistent with this finding, a constitutively active Akt consistently phosphorylated N-CoR leading to its misfolding; while the therapeutic and genetic ablation of Akt largely abrogated the MCDL of N-CoR in AML-M5 cells. Site directed mutagenic analysis of N-CoR identified serine 1450 as the crucial residue whose phosphorylation by Akt was essential for the misfolding and loss of N-CoR protein. Moreover, Akt-induced phosphorylation of N-CoR contributed to the de-repression of Flt3, suggesting a cross talk between Akt signaling and N-CoR misfolding pathway in the pathogenesis of AML-M5. The N-CoR misfolding pathway could be the common downstream thread of pleiotropic Akt signaling activated by various oncogenic insults in some subtypes of leukemia and solid tumors.
核受体共抑制因子(N-CoR)是通用共抑制复合物的关键组成部分,在控制细胞生长和分化中发挥重要作用。正如我们最近所表明的那样,N-CoR 的生长抑制功能在很大程度上依赖于其抑制 Flt3 的能力,Flt3 是正常和恶性造血过程中细胞生长的关键调节剂。我们进一步证明了由于 N-CoR 的错误折叠构象依赖性丧失(MCDL)导致 Flt3 的去抑制如何导致急性髓细胞白血病(AML)中的恶性生长。然而,N-CoR 的 MCDL 的分子机制及其在 AML 发病机制中的意义尚不完全清楚。在这里,我们报告 Akt 诱导的 N-CoR 在公认的 Akt 基序上的磷酸化对于其在 AML(AML-M5)中的错误折叠和随后的丧失至关重要。N-CoR 在几乎所有 AML-M5 衍生细胞中的丝氨酸特异性磷酸化水平明显更高,并且受到 AML-M5 特异性异常蛋白酶活性的处理。为了鉴定与 N-CoR 磷酸化相关的激酶,使用 AML 细胞提取物筛选了一个激活激酶文库;导致鉴定 Akt 为与 N-CoR 磷酸化相关的推定激酶。与这一发现一致,组成型激活的 Akt 一致地磷酸化 N-CoR 导致其错误折叠;而 Akt 的治疗和遗传消融在很大程度上消除了 AML-M5 细胞中 N-CoR 的 MCDL。N-CoR 的定点突变分析鉴定丝氨酸 1450 为关键残基,其磷酸化由 Akt 介导对于 N-CoR 蛋白的错误折叠和丢失是必不可少的。此外,Akt 诱导的 N-CoR 磷酸化有助于 Flt3 的去抑制,表明 Akt 信号传导与 N-CoR 错误折叠途径在 AML-M5 发病机制中的交叉对话。N-CoR 错误折叠途径可能是各种致癌刺激激活的多效性 Akt 信号传导在某些白血病和实体肿瘤亚型中的共同下游线索。