Experimental and Molecular Pathology, Institute of Pathology, Ludwig-Maximilians-Universität München, Thalkirchner Strasse 36, D-80337 Munich, Germany.
Cell Death Dis. 2013 Aug 15;4(8):e775. doi: 10.1038/cddis.2013.282.
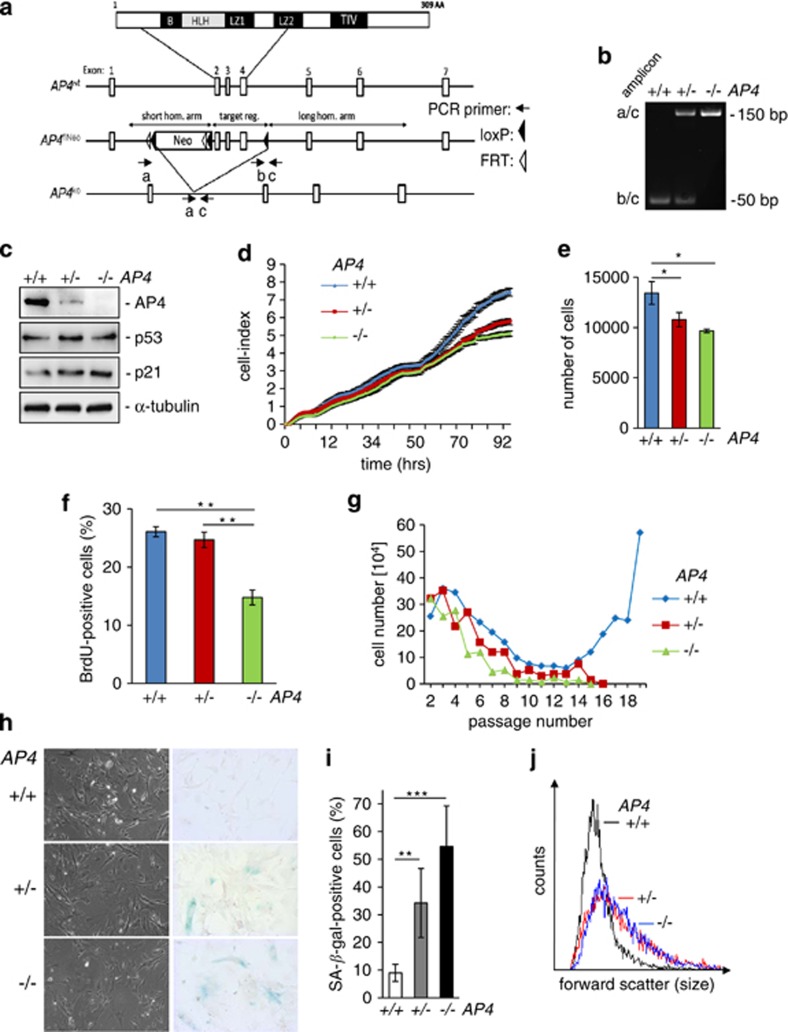
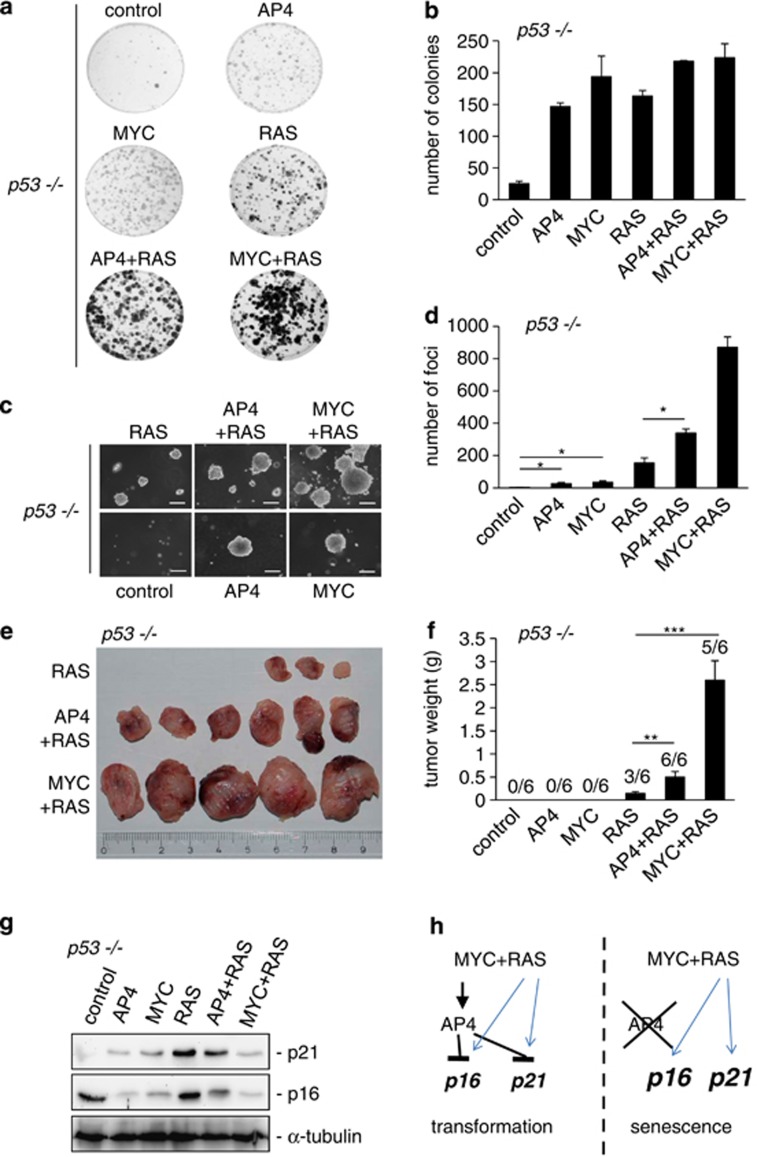
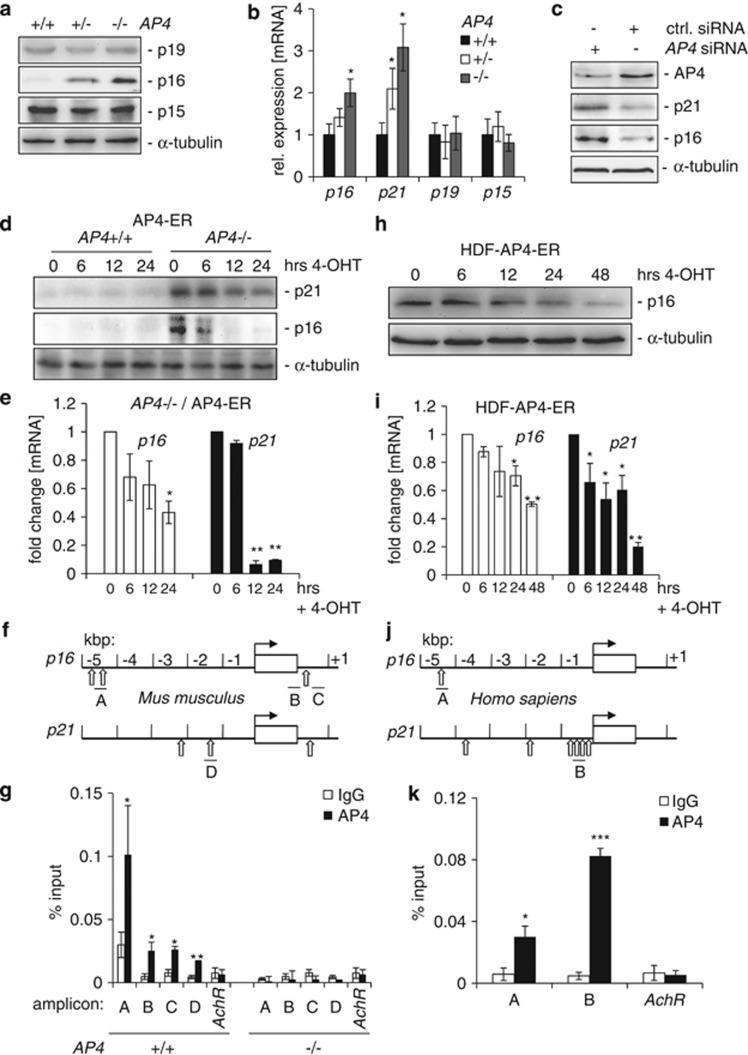
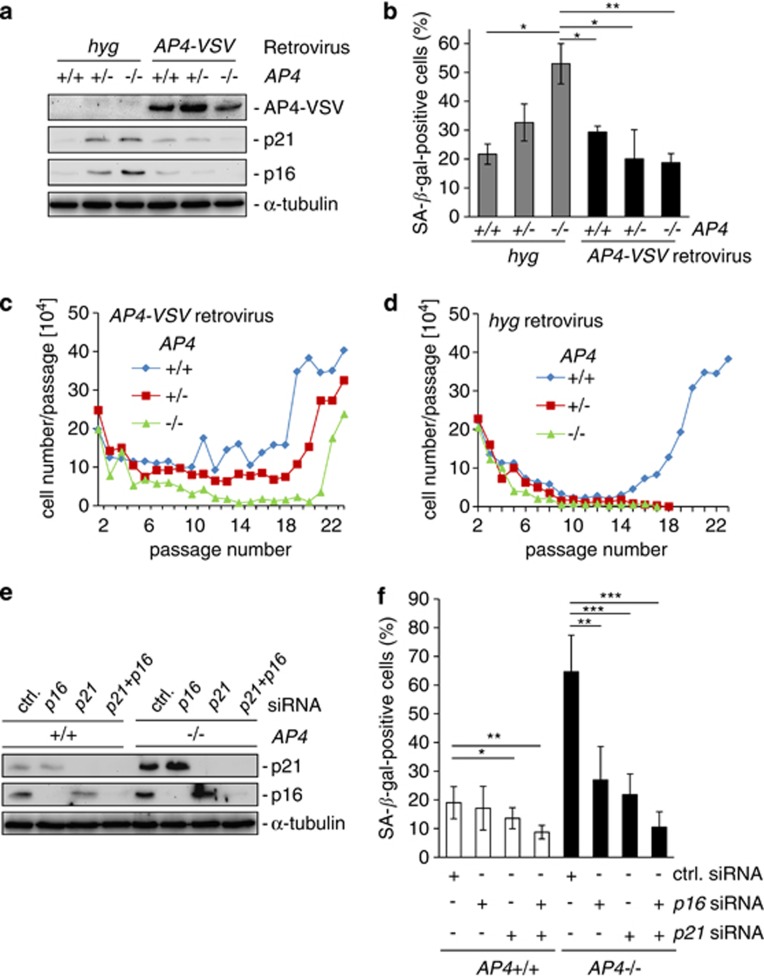
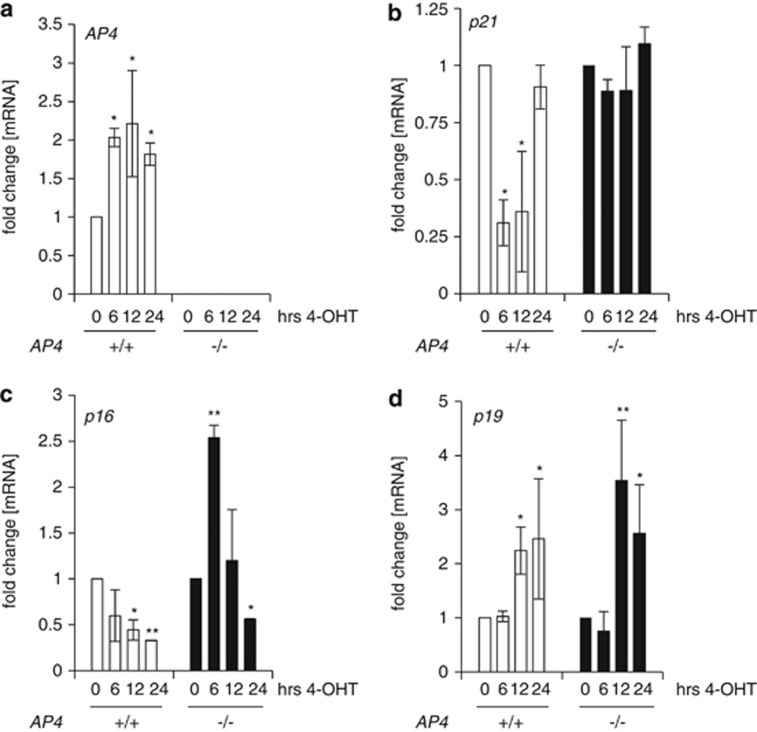
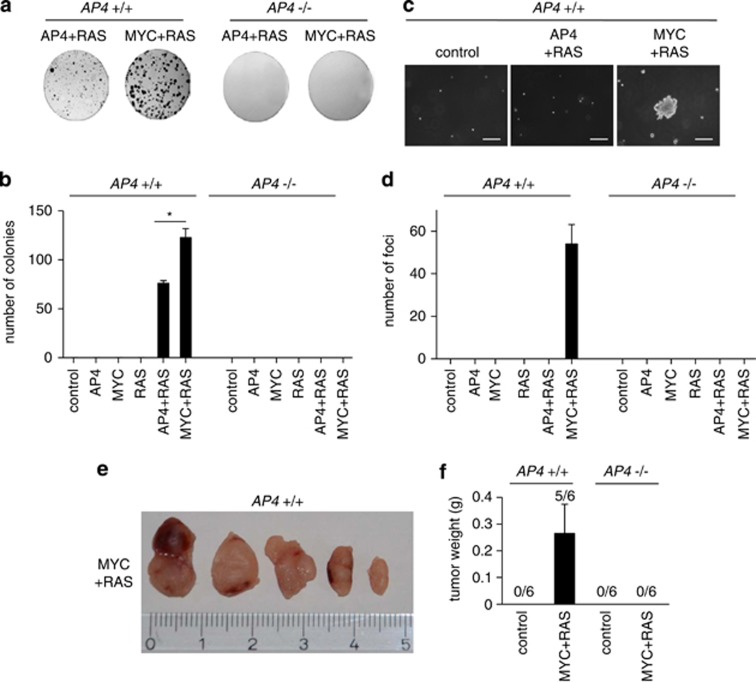
Here we analyzed the function of the c-MYC-inducible basic helix-loop-helix leucine-zipper transcription factor AP4 in AP4-deficient mouse embryo fibroblasts (MEFs). Loss of AP4 resulted in premature senescence and resistance towards immortalization. Senescence was accompanied by induction of the cyclin-dependent kinase inhibitor-encoding genes p16, a known tumor suppressor, and p21, a previously described target for repression by AP4. Notably, AP4 directly repressed p16 expression via conserved E-box motifs in MEFs and human diploid fibroblasts. Senescence caused by AP4-deficiency was prevented by depletion of p16 and/or p21, demonstrating that these factors mediate senescence caused by AP4 loss. As senescence induced by the loss of AP4 was rescued by ectopic AP4, secondary lesions were not involved in causing premature senescence. Activation of c-MYC resulted in repression of p21 and p16 in AP4(+/+), but not in AP4(-/-) MEFs. Furthermore, after combined expression of c-MYC and mutant RAS in MEFs, AP4 was required for colony formation, anchorage-independent growth and tumor formation in mice. In addition, combined ectopic expression of AP4 and mutant RAS in MEFs resulted in colony formation. However, additional loss of the p53 tumor suppressor was necessary for anchorage-independent growth and tumor formation of MEFs by combined AP4 and mutant RAS expression. In conclusion, this study identified AP4 as an oncogenic antagonist of cellular senescence. AP4 achieves this effect by direct repression of p16 and p21, and may thereby critically contribute to c-MYC function and tumor progression.
在这里,我们分析了 c-MYC 诱导的基本螺旋-环-螺旋亮氨酸拉链转录因子 AP4 在 AP4 缺陷型小鼠胚胎成纤维细胞(MEFs)中的功能。AP4 的缺失导致细胞过早衰老和抵抗永生化。衰老伴随着细胞周期蛋白依赖性激酶抑制剂编码基因 p16 的诱导,p16 是一种已知的肿瘤抑制因子,p21 是以前描述的被 AP4 抑制的靶基因。值得注意的是,AP4 通过 MEFs 和人二倍体成纤维细胞中的保守 E 盒基序直接抑制 p16 的表达。AP4 缺失引起的衰老可被 p16 和/或 p21 的耗尽所阻止,表明这些因子介导由 AP4 缺失引起的衰老。由于 AP4 缺失引起的衰老可被异位 AP4 挽救,因此次级病变不参与引起过早衰老。由于 c-MYC 的激活导致 AP4(+/+)中的 p21 和 p16 被抑制,但在 AP4(-/-)的 MEFs 中没有,因此 AP4 的缺失会导致细胞衰老。此外,在 MEFs 中共同表达 c-MYC 和突变型 RAS 后,AP4 对于集落形成、非锚定依赖性生长和在小鼠中形成肿瘤是必需的。此外,在 MEFs 中共同异位表达 AP4 和突变型 RAS 导致集落形成。然而,对于由 AP4 和突变型 RAS 共同表达引起的 MEFs 的非锚定依赖性生长和肿瘤形成,还需要额外丢失肿瘤抑制因子 p53。总之,这项研究确定 AP4 是细胞衰老的致癌拮抗剂。AP4 通过直接抑制 p16 和 p21 来实现这一效应,从而可能对 c-MYC 功能和肿瘤进展做出关键贡献。