Division of Microbiology and Immunology, Pathology Department, University of Utah School of Medicine, Salt Lake City, Utah, United States of America.
PLoS Genet. 2013;9(8):e1003716. doi: 10.1371/journal.pgen.1003716. Epub 2013 Aug 22.
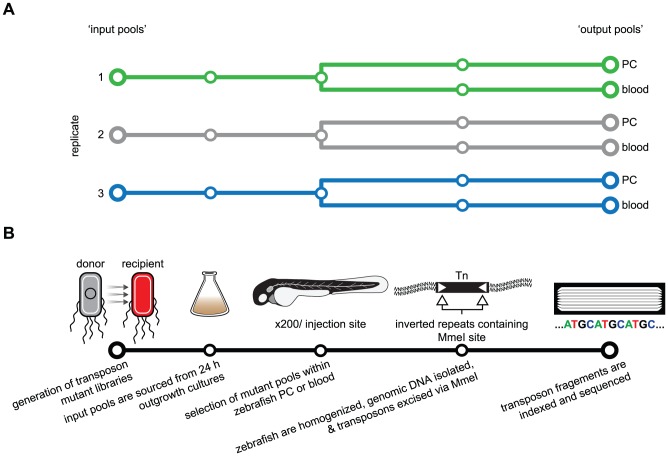
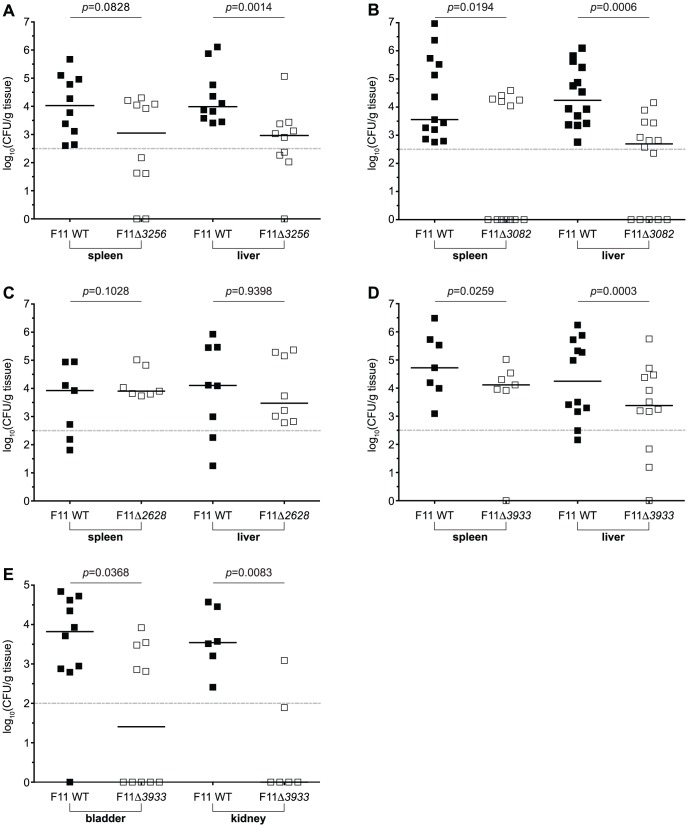
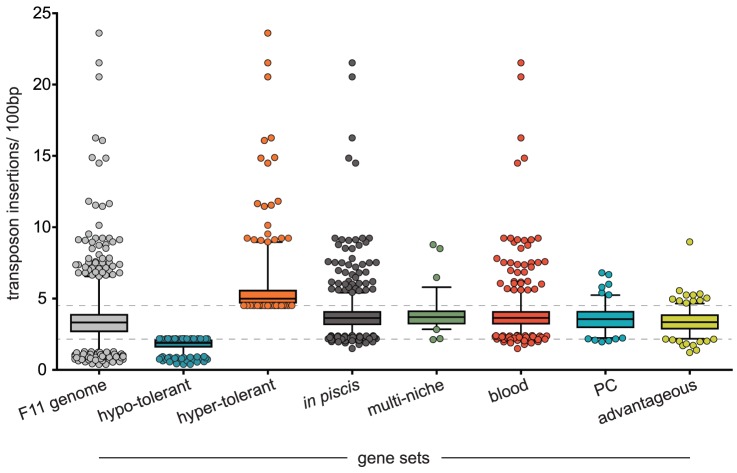
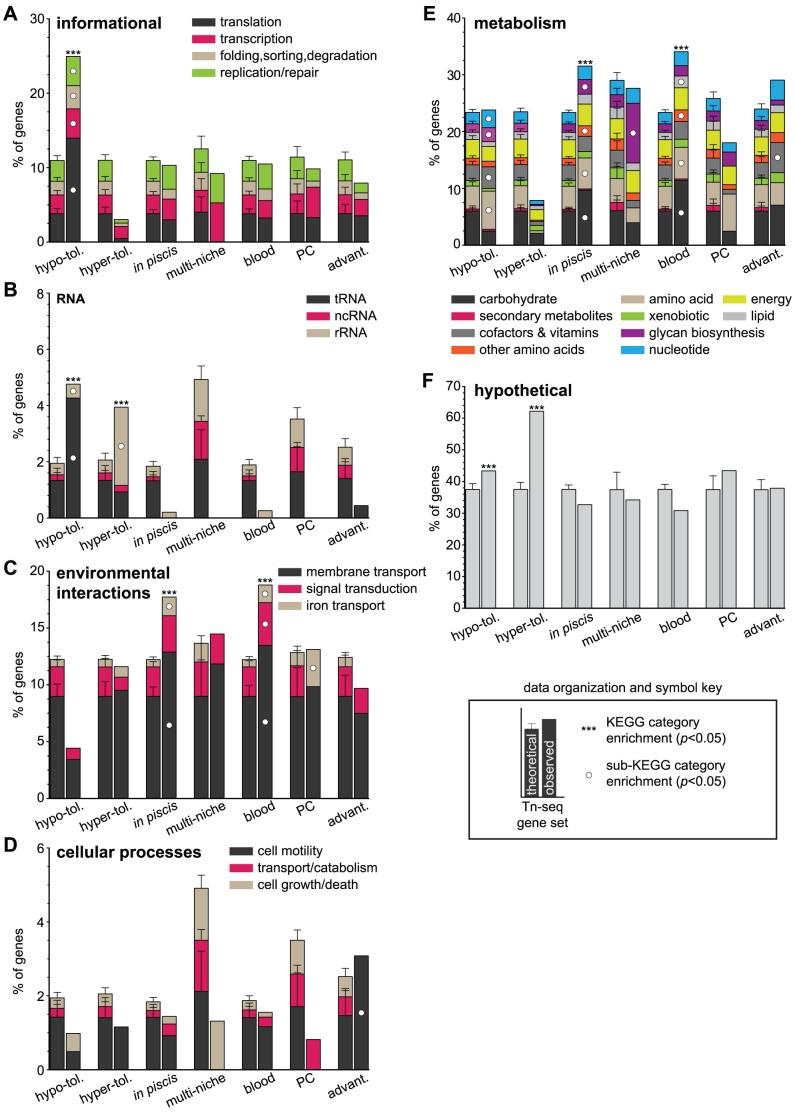
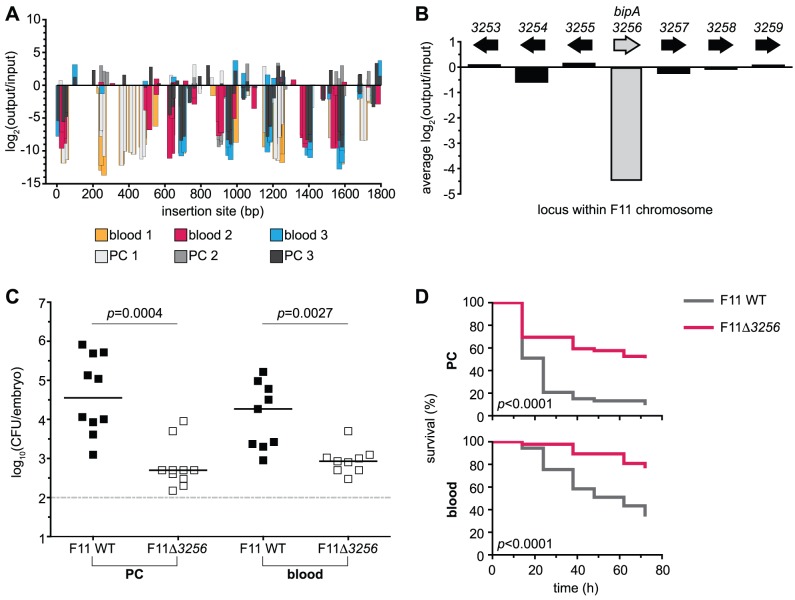
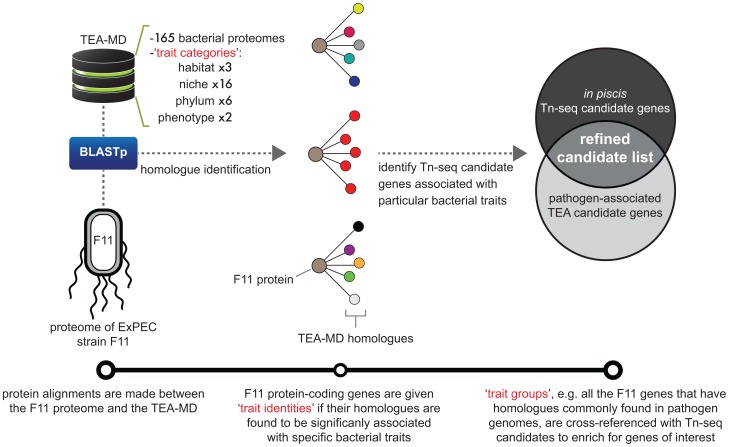
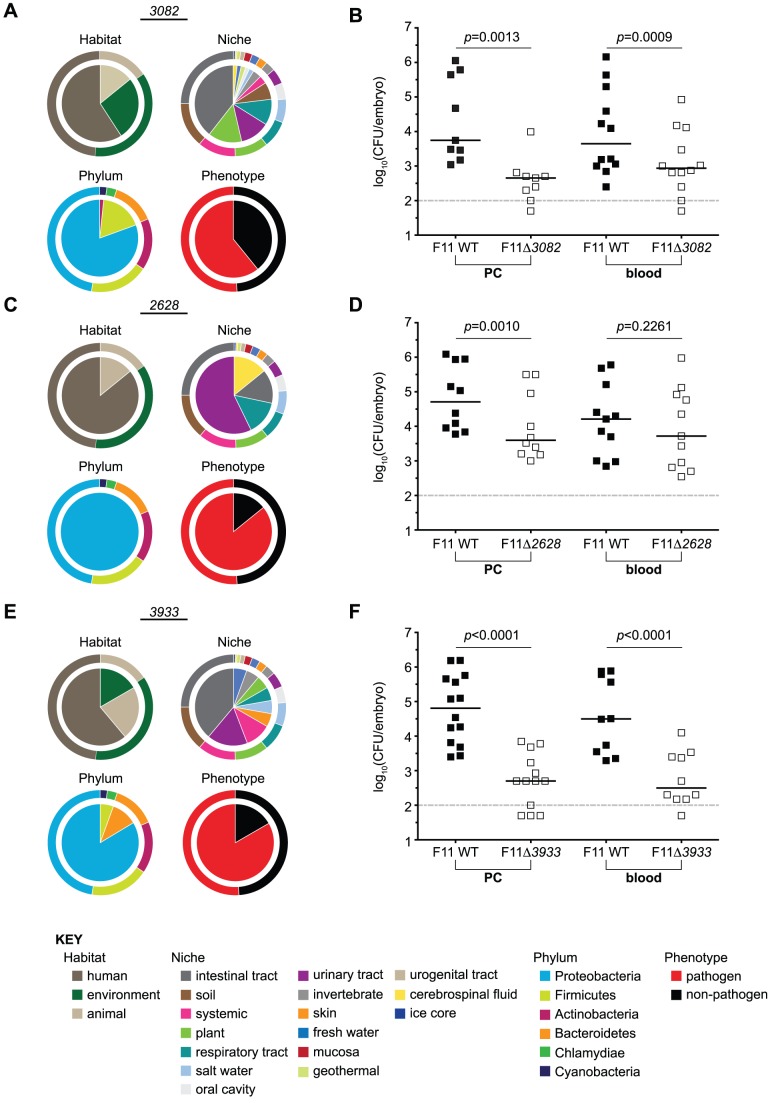
Strains of Extraintestinal Pathogenic Escherichia c oli (ExPEC) exhibit an array of virulence strategies and are a major cause of urinary tract infections, sepsis and meningitis. Efforts to understand ExPEC pathogenesis are challenged by the high degree of genetic and phenotypic variation that exists among isolates. Determining which virulence traits are widespread and which are strain-specific will greatly benefit the design of more effective therapies. Towards this goal, we utilized a quantitative genetic footprinting technique known as transposon insertion sequencing (Tn-seq) in conjunction with comparative pathogenomics to functionally dissect the genetic repertoire of a reference ExPEC isolate. Using Tn-seq and high-throughput zebrafish infection models, we tracked changes in the abundance of ExPEC variants within saturated transposon mutant libraries following selection within distinct host niches. Nine hundred and seventy bacterial genes (18% of the genome) were found to promote pathogen fitness in either a niche-dependent or independent manner. To identify genes with the highest therapeutic and diagnostic potential, a novel Trait Enrichment Analysis (TEA) algorithm was developed to ascertain the phylogenetic distribution of candidate genes. TEA revealed that a significant portion of the 970 genes identified by Tn-seq have homologues more often contained within the genomes of ExPEC and other known pathogens, which, as suggested by the first axiom of molecular Koch's postulates, is considered to be a key feature of true virulence determinants. Three of these Tn-seq-derived pathogen-associated genes--a transcriptional repressor, a putative metalloendopeptidase toxin and a hypothetical DNA binding protein--were deleted and shown to independently affect ExPEC fitness in zebrafish and mouse models of infection. Together, the approaches and observations reported herein provide a resource for future pathogenomics-based research and highlight the diversity of factors required by a single ExPEC isolate to survive within varying host environments.
肠外致病性大肠杆菌(ExPEC)菌株表现出多种毒力策略,是尿路感染、败血症和脑膜炎的主要原因。由于分离株之间存在高度的遗传和表型变异,因此理解 ExPEC 发病机制的努力受到了挑战。确定哪些毒力特征广泛存在,哪些特征是菌株特异性的,将极大地有助于设计更有效的治疗方法。为此,我们利用一种称为转座子插入测序(Tn-seq)的定量遗传足迹技术,并结合比较病原体组学,对参考 ExPEC 分离株的遗传谱进行功能剖析。使用 Tn-seq 和高通量斑马鱼感染模型,我们在不同的宿主小生境中进行选择后,跟踪饱和转座子突变文库中 ExPEC 变体丰度的变化。发现 970 个细菌基因(基因组的 18%)以依赖于小生境或独立的方式促进病原体的适应性。为了确定具有最高治疗和诊断潜力的基因,开发了一种新的Trait Enrichment Analysis(TEA)算法来确定候选基因的系统发育分布。TEA 表明,Tn-seq 鉴定的 970 个基因中有很大一部分具有同源物,这些同源物更常包含在 ExPEC 和其他已知病原体的基因组中,这正如分子 Koch 假设的第一公理所建议的那样,被认为是真正毒力决定因素的关键特征。从 Tn-seq 衍生的三种病原体相关基因——一个转录抑制剂、一个假定的金属内肽酶毒素和一个假定的 DNA 结合蛋白——被删除,并证明它们独立地影响斑马鱼和感染小鼠模型中 ExPEC 的适应性。总之,本文报道的方法和观察结果为未来基于病原体组学的研究提供了资源,并强调了单个 ExPEC 分离株在不同宿主环境中生存所需的因素的多样性。