LOEWE-Center for Synthetic Microbiology (SYNMIKRO), Philipps-Universität Marburg, Hans-Meerwein-Str, 6, D-35043, Marburg, Germany.
BMC Genomics. 2013 Sep 22;14:638. doi: 10.1186/1471-2164-14-638.
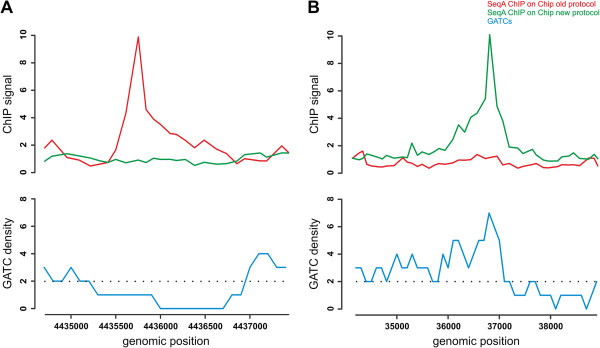
Studies of protein association with DNA on a genome wide scale are possible through methods like ChIP-Chip or ChIP-Seq. Massive problems with false positive signals in our own experiments motivated us to revise the standard ChIP-Chip protocol. Analysis of chromosome wide binding of the alternative sigma factor σ³² in Escherichia coli with this new protocol resulted in detection of only a subset of binding sites found in a previous study by Wade and colleagues. We suggested that the remainder of binding sites detected in the previous study are likely to be false positives. In a recent article the Wade group claimed that our conclusion is wrong and that the disputed sites are genuine σ³² binding sites. They further claimed that the non-detection of these sites in our study was due to low data quality.
RESULTS/DISCUSSION: We respond to the criticism of Wade and colleagues and discuss some general questions of ChIP-based studies. We outline why the quality of our data is sufficient to derive meaningful results. Specific points are: (i) the modifications we introduced into the standard ChIP-Chip protocol do not necessarily result in a low dynamic range, (ii) correlation between ChIP-Chip replicates should not be calculated based on the whole data set as done in transcript analysis, (iii) control experiments are essential for identifying false positives. Suggestions are made how ChIP-based methods could be further optimized and which alternative approaches can be used to strengthen conclusions.
We appreciate the ongoing discussion about the ChIP-Chip method and hope that it helps other scientist to analyze and interpret their results. The modifications we introduced into the ChIP-Chip protocol are a first step towards reducing false positive signals but there is certainly potential for further optimization. The discussion about the σ³² binding sites in question highlights the need for alternative approaches and further investigation of appropriate methods for verification.
通过 ChIP-Chip 或 ChIP-Seq 等方法,可以在全基因组范围内研究蛋白质与 DNA 的结合。我们自己的实验中存在大量的假阳性信号问题,这促使我们修改了标准的 ChIP-Chip 方案。使用该新方案分析大肠杆菌中的替代σ因子σ³²在染色体范围内的结合,结果仅检测到 Wade 及其同事之前研究中发现的结合位点的一个子集。我们认为,之前研究中检测到的其余结合位点可能是假阳性。在最近的一篇文章中,Wade 小组声称我们的结论是错误的,有争议的位点是真正的σ³²结合位点。他们进一步声称,我们的研究中没有检测到这些位点是由于数据质量低。
结果/讨论:我们回应了 Wade 及其同事的批评,并讨论了一些基于 ChIP 的研究的一般性问题。我们概述了为什么我们的数据质量足以得出有意义的结果。具体要点是:(i)我们对标准 ChIP-Chip 方案进行的修改不一定会导致动态范围变低,(ii)ChIP-Chip 重复的相关性不应像在转录分析中那样基于整个数据集进行计算,(iii)对照实验对于识别假阳性至关重要。提出了如何进一步优化基于 ChIP 的方法以及可以使用哪些替代方法来加强结论的建议。
我们感谢对 ChIP-Chip 方法的持续讨论,并希望它有助于其他科学家分析和解释他们的结果。我们引入 ChIP-Chip 方案的修改是减少假阳性信号的第一步,但肯定还有进一步优化的空间。关于有争议的σ³²结合位点的讨论强调了需要替代方法以及进一步研究适当的验证方法的必要性。