Department for Production of Diagnostic Reagents and Research, Blood Transfusion Centre of Slovenia, Šlajmerjeva 6, 1000 Ljubljana, Slovenia.
BMC Neurol. 2013 Sep 25;13:126. doi: 10.1186/1471-2377-13-126.
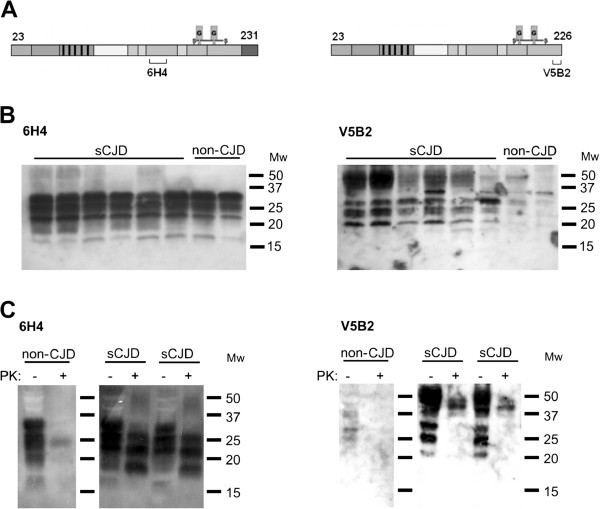
The accumulation of the misfolded forms of cellular prion protein, i.e. prions (PrPSc), in the brain is one of the crucial characteristics of fatal neurodegenerative disorders, called transmissible spongiform encephalopathies (TSEs). Cellular prion protein is normally linked to the cell surface by the glycosylphosphatidylinositol (GPI) anchor. There is accumulating evidence that the GPI-anchorless prion protein may act as an accelerator of formation and propagation of prions. In the TSE affected human brain we have previously discovered a novel GPI-anchorless prion protein fragment, named PrP226*, which ends with the tyrosine 226. This fragment can be labeled specifically by the monoclonal antibody V5B2.
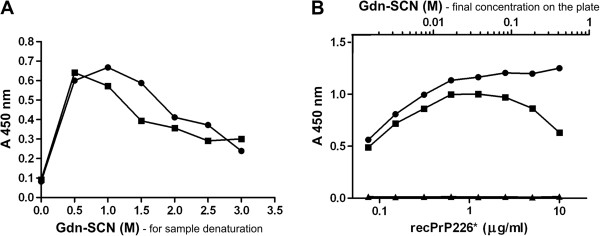
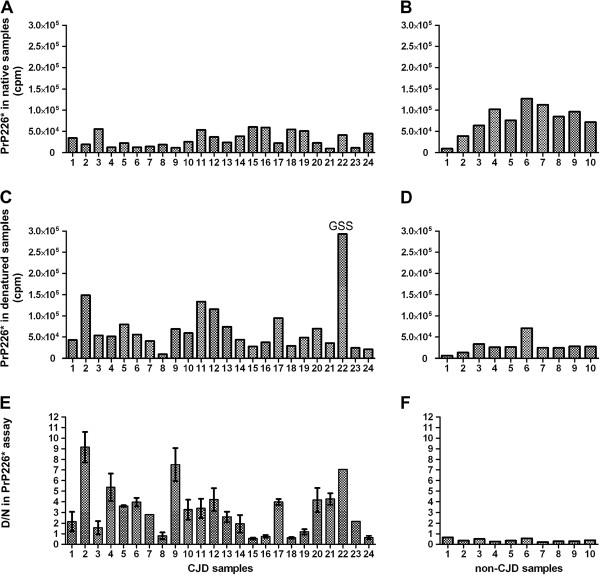
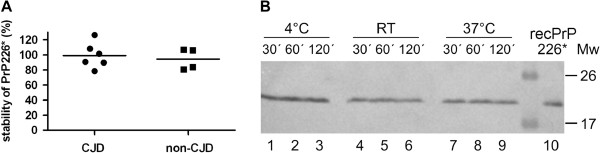
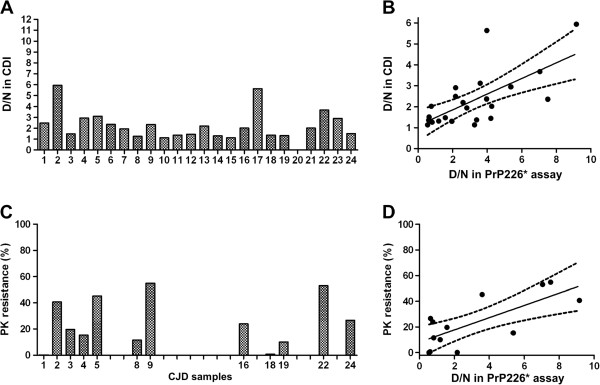
We developed a DELFIA based assay for quick and sensitive detection of the PrP226* fragment in human brain tissue homogenates. By calculating the ratio between the signals of native (N) and denatured (D) samples applied to the assay we were able to observe significant difference between 24 TSE affected brains and 10 control brains. The presence of PrP226* in brain tissue was confirmed by western blot.
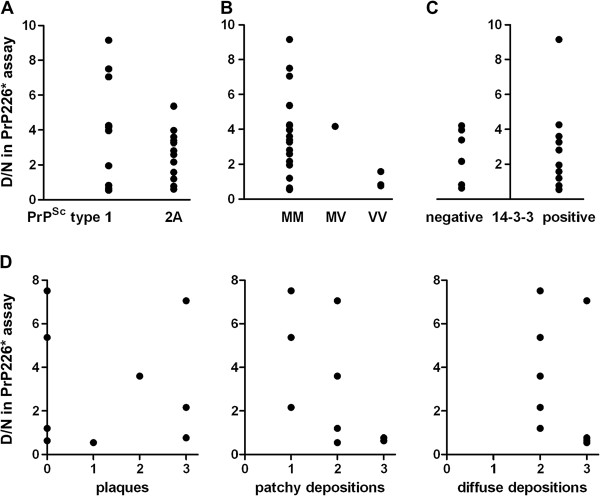
Our results demonstrate that PrP226* is present in small quantities in healthy human brain, whereas in degenerated brain it accumulates in prion aggregates, proportionally to PrPSc. Samples with high D/N ratio generally comprised more proteinase K resistant PrP, while no correlation was found between the quantity of PrP226* and standard classification of Creutzfeldt-Jakob disease (CJD).
In the present study we show that the PrP226* fragment accumulates in prion aggregates and after being released from them by a denaturation procedure, could serve as a proteinase K digestion independent biomarker for human TSEs. The PrP226* assay described in this paper offers a tool to follow and study this unique anchorless PrP fragment in various parts of human brain and possibly also in other tissues and body fluids.
细胞朊病毒蛋白(PrPSc)错误折叠形式的积累是称为传染性海绵状脑病(TSE)的致命神经退行性疾病的关键特征之一。细胞朊病毒蛋白通常通过糖基磷脂酰肌醇(GPI)锚定连接到细胞表面。越来越多的证据表明,无 GPI 锚定的朊病毒蛋白可能作为朊病毒形成和传播的加速剂。在受 TSE 影响的人脑组织中,我们之前发现了一种新型的无 GPI 锚定的朊病毒蛋白片段,命名为 PrP226*,其末端为酪氨酸 226。该片段可以被单克隆抗体 V5B2 特异性标记。
我们开发了一种基于 DELFIA 的测定法,用于快速灵敏地检测人脑组织匀浆中的 PrP226片段。通过计算应用于测定的天然(N)和变性(D)样品的信号比率,我们能够观察到 24 例 TSE 受影响的大脑和 10 例对照大脑之间的显著差异。通过 Western blot 确认了脑组织中 PrP226的存在。
我们的结果表明,PrP226在健康人脑组织中含量较低,而在退化的脑组织中,它与 PrPSc 一起在朊病毒聚集体中积累。具有高 D/N 比值的样品通常包含更多的蛋白水解酶抗性 PrP,而 PrP226的数量与克雅氏病(CJD)的标准分类之间没有相关性。
在本研究中,我们表明 PrP226片段在朊病毒聚集体中积累,并且在用变性程序从聚集体中释放后,可作为人类 TSE 的蛋白酶 K 消化独立的生物标志物。本文描述的 PrP226测定法为研究各种人类脑组织以及可能的其他组织和体液中这种独特的无锚定 PrP 片段提供了一种工具。