Obradors Carla, Echavarren Antonio M
Institute of Chemical Research of Catalonia (ICIQ) , Av. Països Catalans 16, 43007 Tarragona, Spain.
Acc Chem Res. 2014 Mar 18;47(3):902-12. doi: 10.1021/ar400174p. Epub 2013 Oct 31.
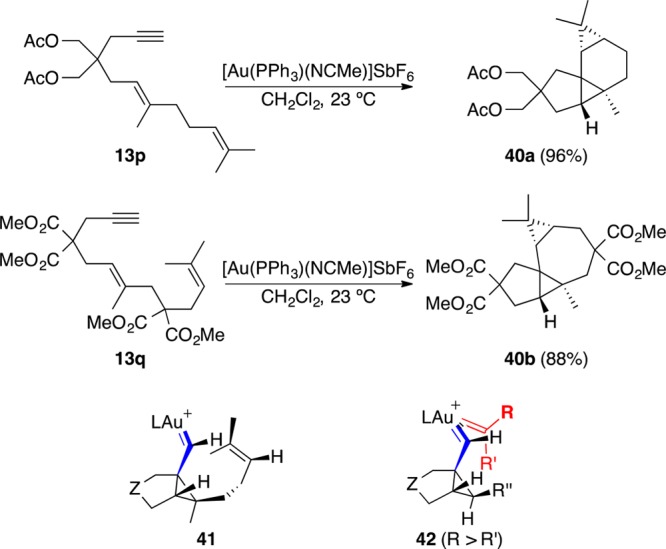
Cycloisomerizations of enynes are probably the most representative carbon-carbon bond forming reactions catalyzed by electrophilic metal complexes. These transformations are synthetically useful because chemists can use them to build complex architectures under mild conditions from readily assembled starting materials. However, these transformations can have complex mechanisms. In general, gold(I) activates alkynes in the presence of any other unsaturated functional group by forming an (η(2)-alkyne)-gold complex. This species reacts readily with nucleophiles, including electron-rich alkenes. In this case, the reaction forms cyclopropyl gold(I) carbene-like intermediates. These can come from different pathways depending on the substitution pattern of the alkyne and the alkene. In the absence of external nucleophiles, 1,n-enynes can form products of skeletal rearrangement in fully intramolecular reactions, which are mechanistically very different from metathesis reactions initiated by the [2 + 2] cycloaddition of a Grubbs-type carbene or other related metal carbenes. In this Account, we discuss how cycloisomerization and addition reactions of substituted enynes, as well as intermolecular reactions between alkynes and alkenes, are best interpreted as proceeding through discrete cationic intermediates in which gold(I) plays a significant role in the stabilization of the positive charge. The most important intermediates are highly delocalized cationic species that some chemists describe as cyclopropyl gold(I) carbenes or gold(I)-stabilized cyclopropylmethyl/cyclobutyl/homoallyl carbocations. However, we prefer the cyclopropyl gold(I) carbene formulation for its simplicity and mnemonic value, highlighting the tendency of these intermediates to undergo cyclopropanation reactions with alkenes. We can add a variety of hetero- and carbonucleophiles to the enynes in the presence of gold(I) in intra- or intermolecular reactions, leading to the corresponding adducts with high stereoselectivity through stereospecific anti-additions. We have also developed stereospecific syn-additions, which probably occur through similar intermediates. The attack of carbonyl groups at the cyclopropyl carbons of the intermediate cyclopropyl gold(I) carbenes initiates a particularly interesting group of reactions. These trigger a cascade transformation that can lead to the formation of two C-C and one C-O bonds. In the fully intramolecular process, this stereospecific transformation has been applied for the synthesis of natural sesquiterpenoids such as (+)-orientalol F and (-)-englerin A. Intra- and intermolecular trapping of cyclopropyl gold(I) carbenes with alkenes leads to the formation of cyclopropanes with significant increase in the molecular complexity, particularly in cases in which this process combines with the migration of propargylic alkoxy and related OR groups. We have recently shown this in the stereoselective total synthesis of the antiviral sesquiterpene (+)-schisanwilsonene by a cyclization/1,5-acetoxy migration/intermolecular cyclopropanation. In this synthesis, the cyclization/1,5-acetoxy migration is faster than the alternative 1,2-acyloxy migration that would result in racemization.

烯炔的环异构化反应可能是亲电金属配合物催化的最具代表性的碳-碳键形成反应。这些转化在合成上很有用,因为化学家可以利用它们在温和条件下从易于组装的起始原料构建复杂的结构。然而,这些转化可能具有复杂的机制。一般来说,金(I)在任何其他不饱和官能团存在下通过形成(η(2)-炔烃)-金配合物来活化炔烃。该物种很容易与亲核试剂反应,包括富电子烯烃。在这种情况下,反应形成环丙基金(I)卡宾样中间体。根据炔烃和烯烃的取代模式,这些中间体可以来自不同的途径。在没有外部亲核试剂的情况下,1,n-烯炔可以在完全分子内反应中形成骨架重排产物,其机理与由格鲁布斯型卡宾或其他相关金属卡宾的[2 + 2]环加成引发的复分解反应有很大不同。在本综述中,我们讨论了取代烯炔的环异构化和加成反应,以及炔烃和烯烃之间的分子间反应,如何最好地解释为通过离散的阳离子中间体进行,其中金(I)在稳定正电荷方面起着重要作用。最重要的中间体是高度离域的阳离子物种,一些化学家将其描述为环丙基金(I)卡宾或金(I)稳定的环丙基甲基/环丁基/高烯丙基碳正离子。然而,我们更喜欢环丙基金(I)卡宾的表述,因为它简单且具有记忆价值,突出了这些中间体与烯烃发生环丙烷化反应的倾向。我们可以在分子内或分子间反应中,在金(I)存在下向烯炔中添加各种杂亲核试剂和碳亲核试剂,通过立体专一的反式加成以高立体选择性得到相应的加合物。我们还开发了立体专一的顺式加成,这可能通过类似的中间体发生。羰基对中间体环丙基金(I)卡宾的环丙基碳的进攻引发了一组特别有趣的反应。这些反应引发了级联转化,可导致形成两个C-C键和一个C-O键。在完全分子内过程中,这种立体专一的转化已应用于天然倍半萜类化合物如(+)-东方醇F和(-)-恩格勒林A的合成。环丙基金(I)卡宾与烯烃的分子内和分子间捕获导致形成环丙烷,分子复杂性显著增加,特别是在该过程与炔丙基烷氧基和相关OR基团的迁移相结合的情况下。我们最近在抗病毒倍半萜(+)-schisanwilsonene的立体选择性全合成中通过环化/1,5-乙酰氧基迁移/分子间环丙烷化展示了这一点。在该合成中,环化/1,5-乙酰氧基迁移比会导致外消旋化的替代1,2-酰氧基迁移更快。