Department of Zoology, University of Oxford, The Tinbergen Building, South Parks Rd, Oxford OX1 3PS, UK.
BMC Evol Biol. 2013 Nov 7;13:243. doi: 10.1186/1471-2148-13-243.
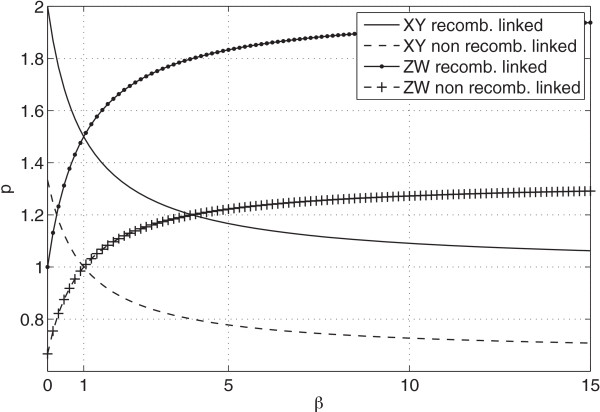
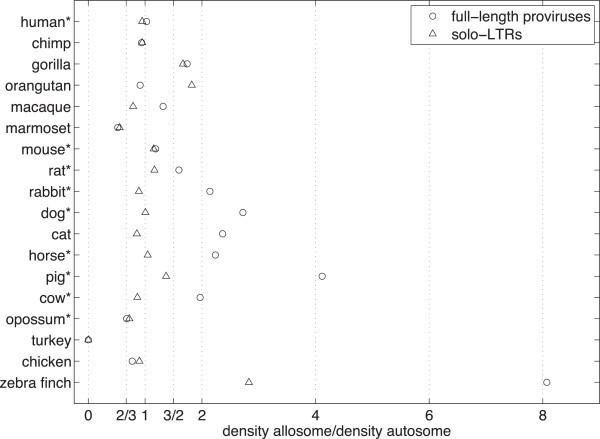
We wish to understand how sex and recombination affect endogenous retroviral insertion and deletion. While theory suggests that the risk of ectopic recombination will limit the accumulation of repetitive DNA in areas of high meiotic recombination, the experimental evidence so far has been inconsistent. Under the assumption of neutrality, we examine the genomes of eighteen species of animal in order to compute the ratio of solo-LTRs that derive from insertions occurring down the male germ line as opposed to the female one (male bias). We also extend the simple idea of comparing autosome to allosome in order to predict the ratio of full-length proviruses we would expect to see under conditions of recombination linked deletion or otherwise.
Using our model, we predict the ratio of allosomal to autosomal full-length proviruses to lie between32 and 23 under increasing male bias in mammals and between 1 and 2 under increasing male bias in birds. In contrast to our expectations, we find that a pattern of male bias is not universal across species and that there is a frequent overabundance of full-length proviruses on the allosome beyond the ratios predicted by our model.
We use our data as a whole to argue that full-length proviruses should be treated as deleterious mutations or as effectively neutral mutations whose persistence in a full-length state is linked to the rate of meiotic recombination and whose origin is not universally male biased. These conclusions suggest that retroviral insertions on the allosome may be more prolific and that it might be possible to identify mechanisms of replication that are enhanced in the female sex.
我们希望了解性别和重组如何影响内源性逆转录病毒的插入和缺失。虽然理论表明,异位重组的风险将限制高减数分裂重组区域中重复 DNA 的积累,但到目前为止,实验证据并不一致。在中性假设下,我们检查了十八种动物的基因组,以计算源自雄性生殖系而非雌性生殖系插入的 solo-LTR 与后者之比(雄性偏向)。我们还扩展了将常染色体与异染色体进行比较的简单想法,以预测在重组连锁缺失或其他情况下我们预计会看到的全长前病毒的比例。
使用我们的模型,我们预测在哺乳动物中雄性偏向增加的情况下,全染色体与常染色体全长前病毒的比例介于 32 到 23 之间,在鸟类中雄性偏向增加的情况下,该比例介于 1 到 2 之间。与我们的预期相反,我们发现雄性偏向的模式并非在所有物种中普遍存在,并且全染色体上全长前病毒的过度丰度经常超过我们模型预测的比例。
我们将我们的数据作为一个整体来论证,全长前病毒应被视为有害突变或有效中性突变,其在全长状态下的持续存在与减数分裂重组率有关,其起源并非普遍雄性偏向。这些结论表明,异染色体上的逆转录病毒插入可能更丰富,并且可能有可能识别出在雌性中增强的复制机制。