Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 50 Ilwon-dong Gangnam-Gu, Seoul 135-710, Korea.
BMC Med Genet. 2013 Dec 5;14:125. doi: 10.1186/1471-2350-14-125.
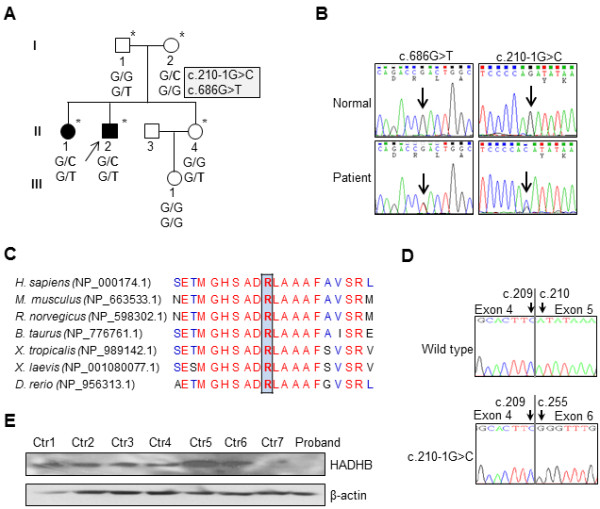
Charcot-Marie-Tooth disease (CMT) is a heterogeneous disorder of the peripheral nervous system. So far, mutations in hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), beta subunit (HADHB) gene exhibit three distinctive phenotypes: severe neonatal presentation with cardiomyopathy, hepatic form with recurrent hypoketotic hypoglycemia, and later-onset axonal sensory neuropathy with episodic myoglobinuria.
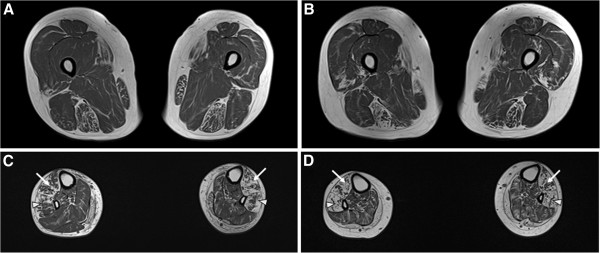
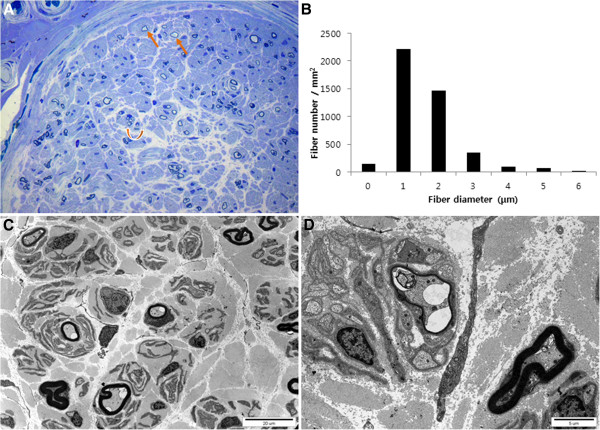
To identify the causative and characterize clinical features of a Korean family with motor and sensory neuropathies, whole exome study (WES), histopathologic study of distal sural nerve, and lower limb MRIs were performed.
WES revealed that a compound heterozygous mutation in HADHB is the causative of the present patients. The patients exhibited an early-onset axonal sensorimotor neuropathy without episodic myoglobinuria, and showed typical clinical and electrophysiological features of CMT including predominant distal muscle weakness and atrophy. Histopathologic findings of sural nerve were compatible with an axonal CMT neuropathy. Furthermore, they didn't exhibit any other symptoms of the previously reported HADHB patients.
These data implicate that mutation in HADHB gene can also cause early-onset axonal CMT instead of typical manifestations in mitochondrial trifunctional protein (MTP) deficiency. Therefore, this study is the first report of a new subtype of autosomal recessive axonal CMT by a compound heterozygous mutation in HADHB, and will expand the clinical and genetic spectrum of HADHB.
Charcot-Marie-Tooth 病(CMT)是一种周围神经系统的异质性疾病。到目前为止,羟酰基辅酶 A 脱氢酶/3-酮酰基辅酶 A 硫解酶/烯酰基辅酶 A 水合酶(三功能蛋白)β亚基(HADHB)基因的突变表现出三种不同的表型:伴有心肌病的严重新生儿期表现、伴有复发性低酮性低血糖的肝型和迟发性轴索性感觉神经病伴阵发性肌红蛋白尿。
为了确定一个有运动和感觉神经病的韩国家族的致病原因并描述其临床特征,进行了全外显子组研究(WES)、远端腓肠神经组织病理学研究和下肢 MRI。
WES 显示 HADHB 的复合杂合突变是本患者的致病原因。患者表现为早发性轴索性感觉运动神经病,无阵发性肌红蛋白尿,表现为 CMT 的典型临床和电生理特征,包括主要的远端肌肉无力和萎缩。腓肠神经的组织病理学发现与轴索性 CMT 神经病相符。此外,他们没有表现出先前报道的 HADHB 患者的任何其他症状。
这些数据表明 HADHB 基因突变也可引起早发性轴索性 CMT,而不是线粒体三功能蛋白(MTP)缺乏症的典型表现。因此,本研究首次报道了由 HADHB 复合杂合突变引起的新的常染色体隐性轴索性 CMT 亚型,并将扩展 HADHB 的临床和遗传谱。