Poulter James A, Brookes Steven J, Shore Roger C, Smith Claire E L, Abi Farraj Layal, Kirkham Jennifer, Inglehearn Chris F, Mighell Alan J
Leeds Institutes of Molecular Medicine, University of Leeds, Leeds LS9 7TF, UK.
Hum Mol Genet. 2014 Apr 15;23(8):2189-97. doi: 10.1093/hmg/ddt616. Epub 2013 Dec 6.
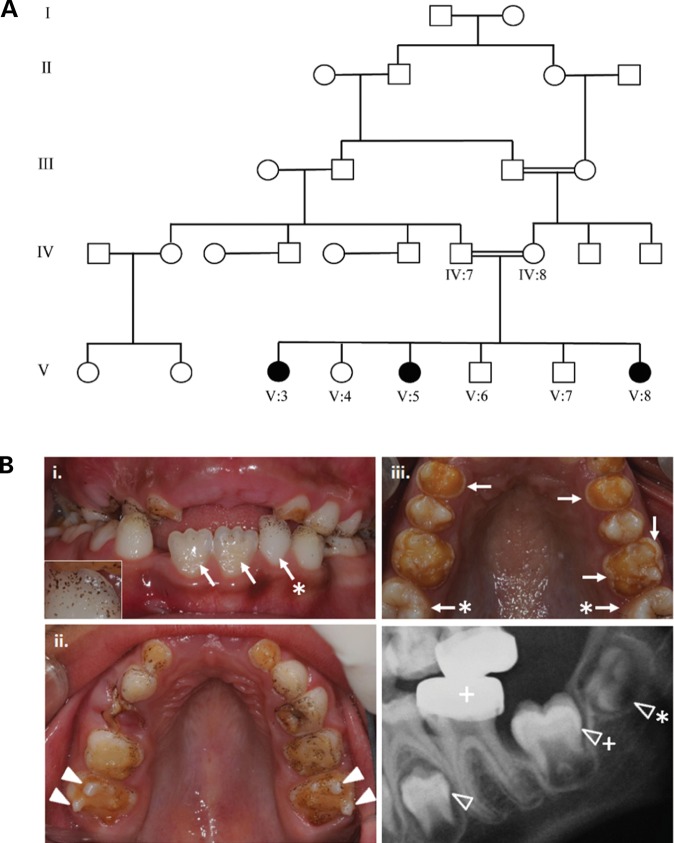
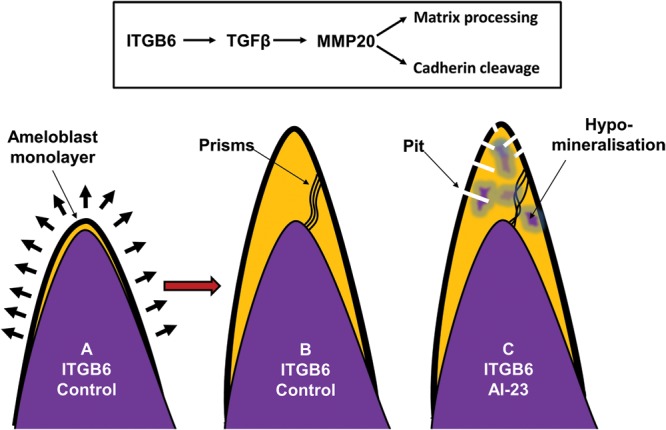
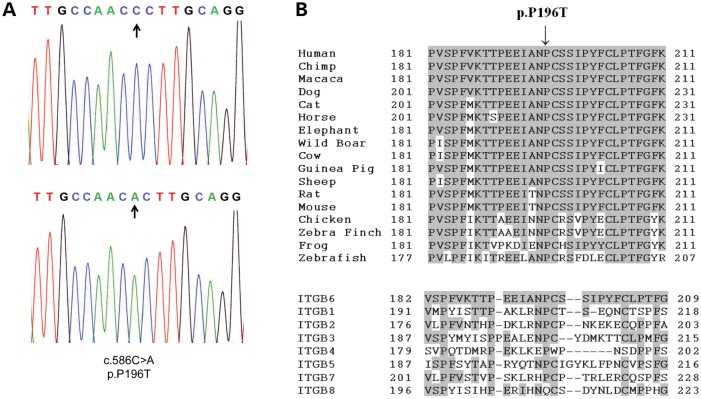
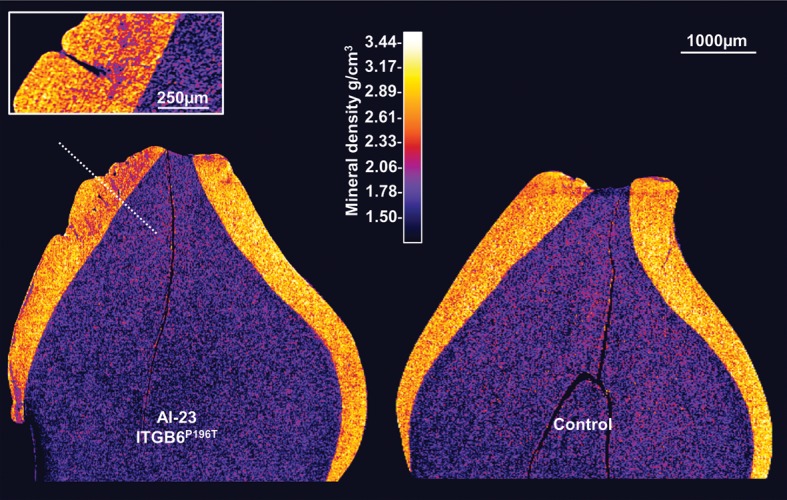
We identified a family in which pitted hypomineralized amelogenesis imperfecta (AI) with premature enamel failure segregated in an autosomal recessive fashion. Whole-exome sequencing revealed a missense mutation (c.586C>A, p.P196T) in the I-domain of integrin-β6 (ITGB6), which is consistently predicted to be pathogenic by all available programmes and is the only variant that segregates with the disease phenotype. Furthermore, a recent study revealed that mice lacking a functional allele of Itgb6 display a hypomaturation AI phenotype. Phenotypic characterization of affected human teeth in this study showed areas of abnormal prismatic organization, areas of low mineral density and severe abnormal surface pitting in the tooth's coronal portion. We suggest that the pathogenesis of this form of AI may be due to ineffective ligand binding of ITGB6 resulting in either compromised cell-matrix interaction or compromised ITGB6 activation of transforming growth factor-β (TGF-β) impacting indirectly on ameloblast-ameloblast interactions and proteolytic processing of extracellular matrix proteins via MMP20. This study adds to the list of genes mutated in AI and further highlights the importance of cell-matrix interactions during enamel formation.
我们鉴定出一个家族,其中伴有釉质过早缺失的凹陷性矿化不全型牙釉质发育不全(AI)以常染色体隐性方式遗传。全外显子组测序显示整合素β6(ITGB6)的I结构域存在一个错义突变(c.586C>A,p.P196T),所有现有程序均一致预测该突变具有致病性,且是唯一与疾病表型共分离的变异。此外,最近一项研究表明,缺乏功能性Itgb6等位基因的小鼠表现出成熟不全型AI表型。本研究中对受累人类牙齿的表型特征分析显示,牙冠部分存在棱柱体组织异常区域、低矿物质密度区域以及严重的表面异常凹陷。我们认为,这种形式的AI发病机制可能是由于ITGB6的配体结合无效,导致细胞-基质相互作用受损,或ITGB6对转化生长因子-β(TGF-β)的激活受损,进而通过基质金属蛋白酶20间接影响成釉细胞-成釉细胞相互作用以及细胞外基质蛋白的蛋白水解过程。本研究增加了在AI中发生突变的基因列表,并进一步突出了牙釉质形成过程中细胞-基质相互作用的重要性。