Iemolo Francesco, Pizzo Federica, Albeggiani Giuseppe, Zizzo Carmela, Colomba Paolo, Scalia Simone, Bartolotta Caterina, Duro Giovanni
CNR-IBIM: Institute of Biomedicine and Molecular Immunology "A, Monroy", Via Ugo la Malfa n,153, Palermo, Italy.
BMC Res Notes. 2014 Jan 7;7:11. doi: 10.1186/1756-0500-7-11.
Fabry disease is an X-linked inherited metabolic condition where the deficit of the α-galactosidase A enzyme, encoded by the GLA gene, leads to glycosphingolipid storage, mainly globotriaosylceramide. To date, more than 600 mutations have been identified in human GLA gene that are responsible for FD, including missense and nonsense mutations, small and large deletions. Such mutations are usually inherited, and cases of de novo onset occur rarely.
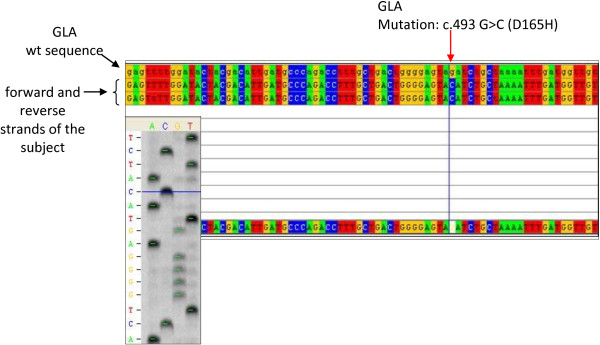
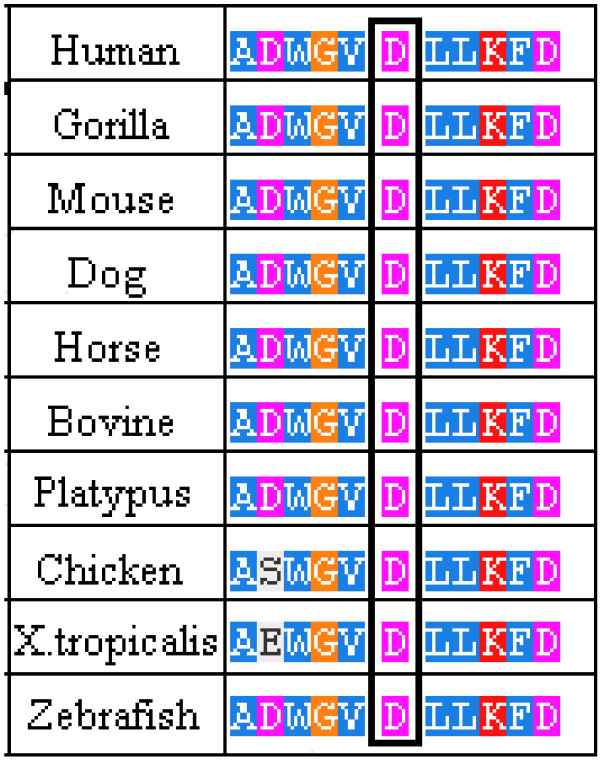
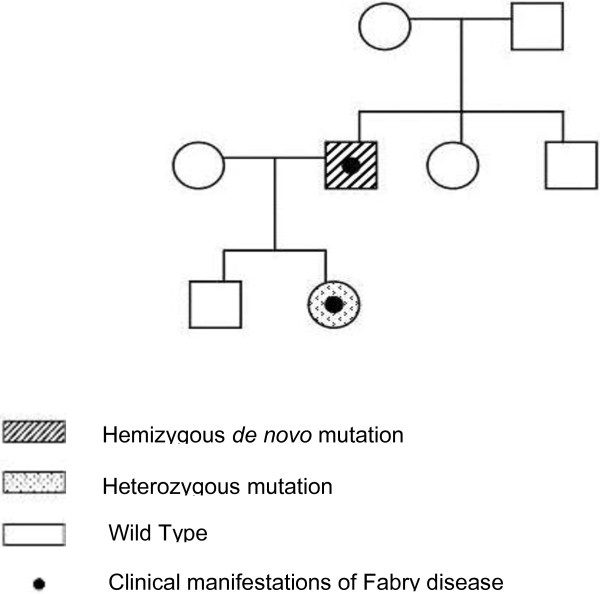
In this article we report an interesting case of a 44-year-old male patient suffering from a severe form of Fabry disease, with negative family history. The patient showed signs such as cornea verticillata, angiokeratomas, cardiac and neurological manifestations, an end-stage renal disease and he had low α-galactosidase A activity. We detected, in this subject, the mutation c.493 G > C in the third exon of the GLA gene which causes the amino acid substitution D165H in the protein. This mutation affects the amino acid - belonging to the group of buried residues - involved, probably, in the preservation of the protein folding. Moreover, studies of multiple sequence alignment indicate that this amino acid is highly conserved, thus strengthening the hypothesis that it is a key amino acid to the enzyme functionality.The study of the relatives of the patient showed that, surprisingly, none of the members of his family of origin had this genetic alteration, suggesting a de novo mutation. Only his 11-year-old daughter - showing acroparaesthesias and heat intolerance with reduced enzymatic activity - had the same mutation.
We suggest that a non-inherited mutation of the α-galactosidase A gene is responsible for Fabry disease in the patient who had reduced enzyme activity and classical clinical manifestations of the disease. In a family, it is rare to find only one Fabry disease affected subject with a de novo mutation. These findings emphasize the importance of early diagnosis, genetic counselling, studying the genealogical tree of the patients and starting enzyme replacement therapy to prevent irreversible vital organ damage that occurs during the course of the disease.
法布里病是一种X连锁遗传性代谢疾病,由GLA基因编码的α-半乳糖苷酶A酶缺乏导致糖鞘脂蓄积,主要是球三糖神经酰胺。迄今为止,已在人类GLA基因中鉴定出600多种导致法布里病的突变,包括错义突变和无义突变、小缺失和大缺失。这些突变通常是遗传的,新发病例很少见。
在本文中,我们报告了一例有趣的病例,一名44岁男性患者患有严重形式的法布里病,家族史阴性。患者表现出角膜涡状浑浊、血管角质瘤、心脏和神经症状、终末期肾病等体征,且α-半乳糖苷酶A活性较低。我们在该患者中检测到GLA基因第三外显子中的c.493 G > C突变,该突变导致蛋白质中的氨基酸替换D165H。此突变影响属于埋藏残基组的氨基酸,可能参与蛋白质折叠的维持。此外,多序列比对研究表明该氨基酸高度保守,从而强化了它是酶功能关键氨基酸的假设。对患者亲属的研究表明,令人惊讶的是,其原生家庭的成员均无此基因改变,提示为新发突变。只有他11岁的女儿表现出肢端感觉异常和不耐热,酶活性降低,有相同的突变。
我们认为,α-半乳糖苷酶A基因的非遗传性突变导致了该患者酶活性降低并出现该疾病的典型临床表现。在一个家族中,仅发现一名患有新发突变的法布里病患者的情况很少见。这些发现强调了早期诊断、遗传咨询、研究患者家谱以及开始酶替代治疗以预防疾病过程中发生不可逆重要器官损害的重要性。