Kelberman Daniel, Islam Lily, Lakowski Jörn, Bacchelli Chiara, Chanudet Estelle, Lescai Francesco, Patel Aara, Stupka Elia, Buck Anja, Wolf Stephan, Beales Philip L, Jacques Thomas S, Bitner-Glindzicz Maria, Liasis Alki, Lehmann Ordan J, Kohlhase Jürgen, Nischal Ken K, Sowden Jane C
Ulverscroft Vision Research Group.
Hum Mol Genet. 2014 May 15;23(10):2511-26. doi: 10.1093/hmg/ddt643. Epub 2014 Jan 9.
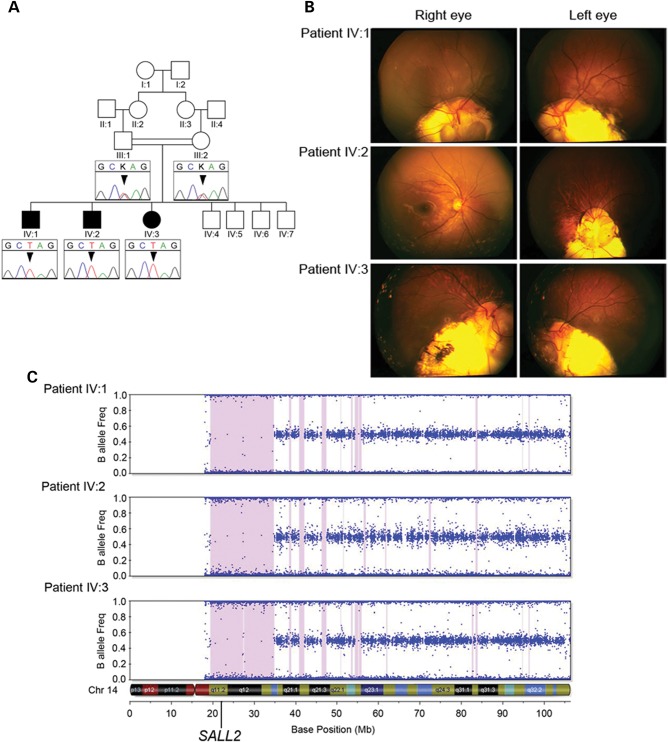
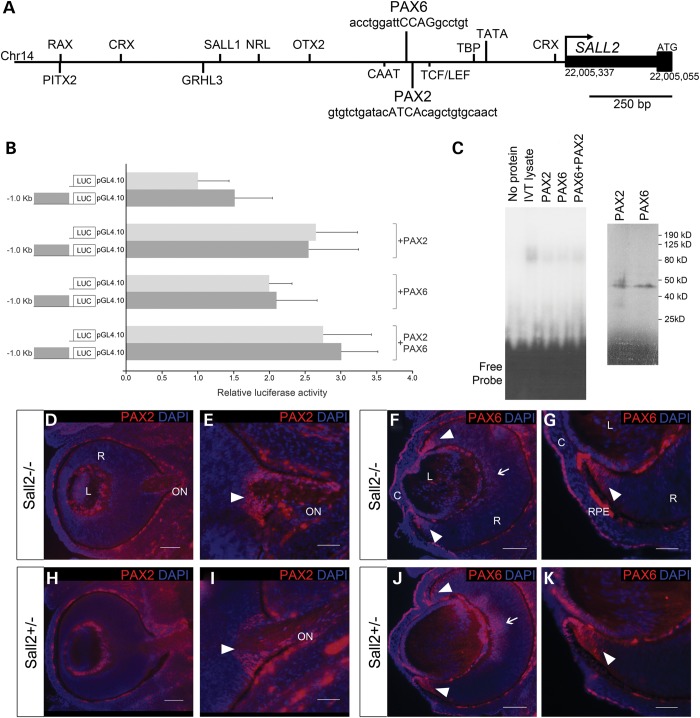
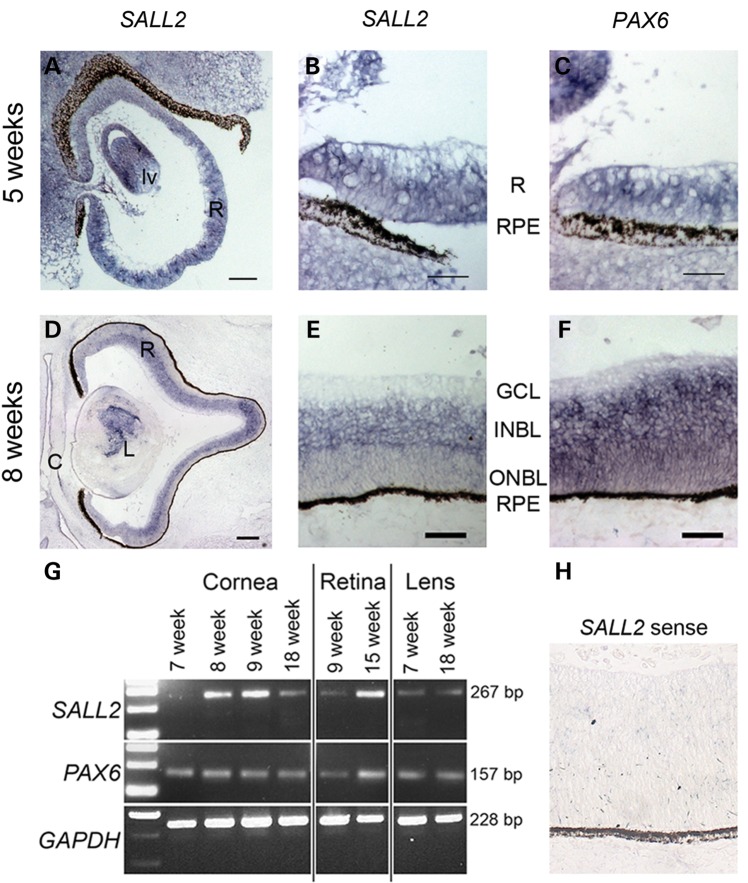
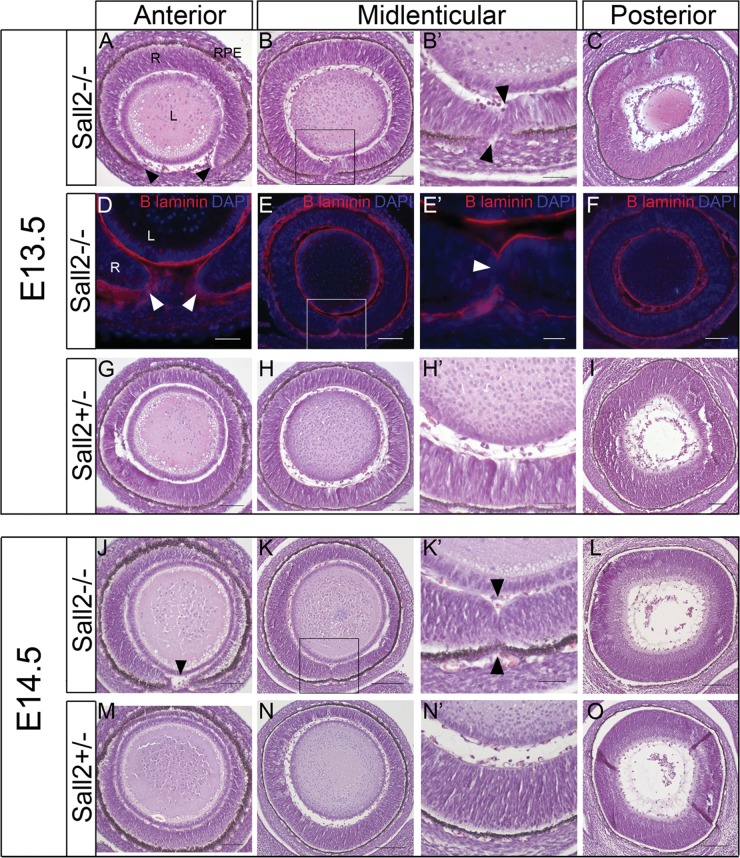
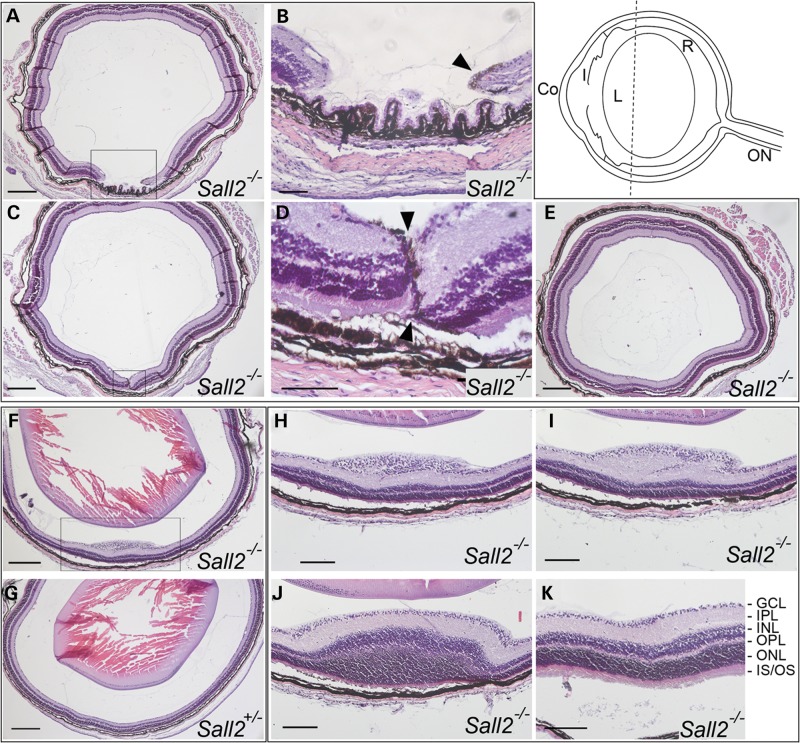
Ocular coloboma is a congenital defect resulting from failure of normal closure of the optic fissure during embryonic eye development. This birth defect causes childhood blindness worldwide, yet the genetic etiology is poorly understood. Here, we identified a novel homozygous mutation in the SALL2 gene in members of a consanguineous family affected with non-syndromic ocular coloboma variably affecting the iris and retina. This mutation, c.85G>T, introduces a premature termination codon (p.Glu29*) predicted to truncate the SALL2 protein so that it lacks three clusters of zinc-finger motifs that are essential for DNA-binding activity. This discovery identifies SALL2 as the third member of the Drosophila homeotic Spalt-like family of developmental transcription factor genes implicated in human disease. SALL2 is expressed in the developing human retina at the time of, and subsequent to, optic fissure closure. Analysis of Sall2-deficient mouse embryos revealed delayed apposition of the optic fissure margins and the persistence of an anterior retinal coloboma phenotype after birth. Sall2-deficient embryos displayed correct posterior closure toward the optic nerve head, and upon contact of the fissure margins, dissolution of the basal lamina occurred and PAX2, known to be critical for this process, was expressed normally. Anterior closure was disrupted with the fissure margins failing to meet, or in some cases misaligning leading to a retinal lesion. These observations demonstrate, for the first time, a role for SALL2 in eye morphogenesis and that loss of function of the gene causes ocular coloboma in humans and mice.
眼裂缺是胚胎眼发育过程中视裂正常闭合失败导致的先天性缺陷。这种出生缺陷在全球范围内导致儿童失明,但其遗传病因却知之甚少。在此,我们在一个近亲家庭的成员中鉴定出SALL2基因的一个新的纯合突变,该家庭患有非综合征性眼裂缺,可不同程度地影响虹膜和视网膜。这个突变,c.85G>T,引入了一个过早的终止密码子(p.Glu29*),预计会截断SALL2蛋白,使其缺少对DNA结合活性至关重要的三簇锌指基序。这一发现确定SALL2是果蝇同源异形Spalt样发育转录因子基因家族中第三个与人类疾病相关的成员。SALL2在视裂闭合时以及闭合后在发育中的人类视网膜中表达。对Sall2缺陷小鼠胚胎的分析显示,视裂边缘的并置延迟,出生后视网膜前裂缺表型持续存在。Sall2缺陷胚胎在朝向视神经头的方向上显示出正确的后部闭合,并且在裂缘接触时,基膜溶解发生,已知对此过程至关重要的PAX2正常表达。前部闭合受到破坏,裂缘未能会合,或者在某些情况下错位,导致视网膜病变。这些观察结果首次证明了SALL2在眼睛形态发生中的作用,并且该基因功能丧失会导致人类和小鼠出现眼裂缺。