Medical Microbiology, Department of Laboratory Medicine Malmö, Lund University, Jan Waldenströms gata 59, SE-205 02 Malmö, Sweden.
BMC Genomics. 2014 Jan 18;15(1):38. doi: 10.1186/1471-2164-15-38.
The incidence of invasive disease caused by encapsulated Haemophilus influenzae type f (Hif) has increased in the post-H. influenzae type b (Hib) vaccine era. We previously annotated the first complete Hif genome from a clinical isolate (KR494) that caused septic shock and necrotizing myositis. Here, the full genome of Hif KR494 was compared to sequenced reference strains Hib 10810, capsule type d (Hid) Rd Kw20, and finally nontypeable H. influenzae 3655. The goal was to identify possible genomic characteristics that may shed light upon the pathogenesis of Hif.
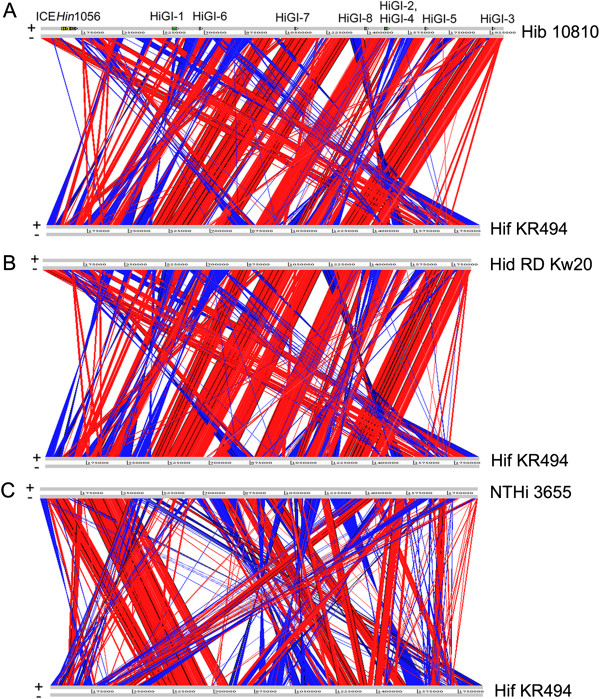
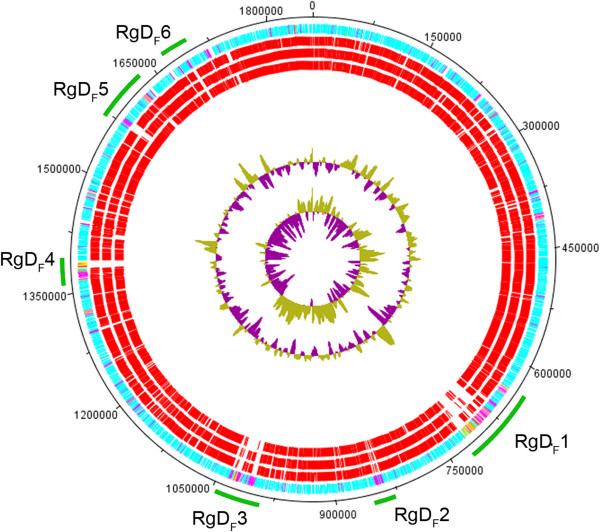
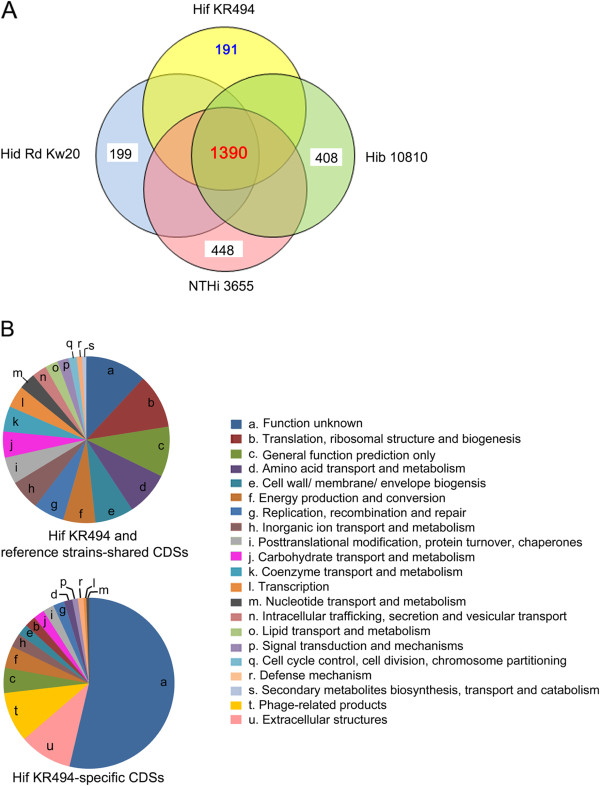
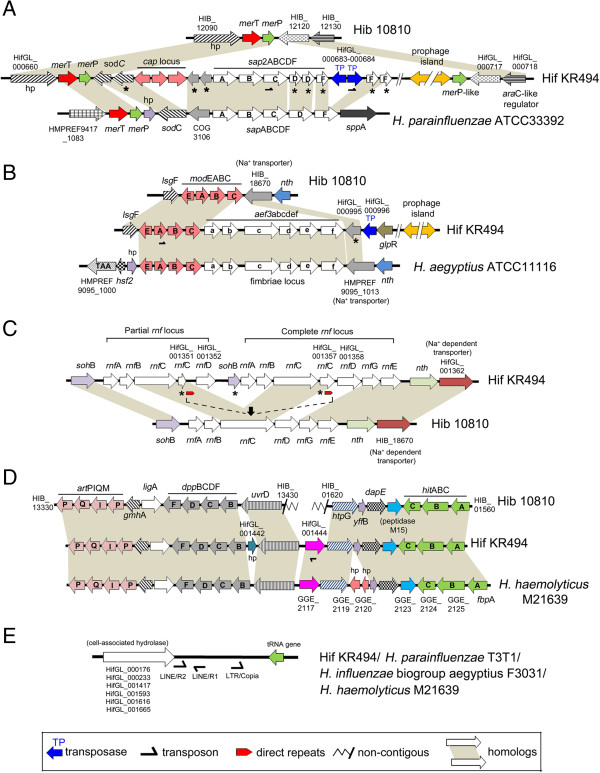
The Hif KR494 genome exhibited large regions of synteny with other H. influenzae, but also distinct genome rearrangements. A predicted Hif core genome of 1390 genes was shared with the reference strains, and 6 unique genomic regions comprising half of the 191 unique coding sequences were revealed. The majority of these regions were inserted genetic fragments, most likely derived from the closely-related Haemophilus spp. including H. aegyptius, H. haemolyticus and H. parainfluenzae. Importantly, the KR494 genome possessed several putative virulence genes that were distinct from non-type f strains. These included the sap2 operon, aef3 fimbriae, and genes for kanamycin nucleotidyltranserase, iron-utilization proteins, and putative YadA-like trimeric autotransporters that may increase the bacterial virulence. Furthermore, Hif KR494 lacked a hisABCDEFGH operon for de novo histidine biosynthesis, hmg locus for lipooligosaccharide biosynthesis and biofilm formation, the Haemophilus antibiotic resistance island and a Haemophilus secondary molybdate transport system. We confirmed the histidine auxotrophy and kanamycin resistance in Hif by functional experiments. Moreover, the pattern of unique or missing genes of Hif KR494 was similar in 20 Hif clinical isolates obtained from different years and geographical areas. A cross-species comparison revealed that the Hif genome shared more characteristics with H. aegyptius than Hid and NTHi.
The genomic comparative analyses facilitated identification of genotypic characteristics that may be related to the specific virulence of Hif. In relation to non-type f H. influenzae strains, the Hif genome contains differences in components involved in metabolism and survival that may contribute to its invasiveness.
荚膜性流感嗜血杆菌 f 型(Hif)引起的侵袭性疾病的发病率在乙型流感嗜血杆菌(Hib)疫苗时代后有所增加。我们之前对引起感染性休克和坏死性肌炎的临床分离株 KR494 进行了 Hif 全基因组注释。在此,我们将 Hif KR494 的全基因组与已测序的参考株 Hib 10810、荚膜型 d(Hid)Rd Kw20 以及最后一种非典型流感嗜血杆菌 3655 进行比较。目的是确定可能阐明 Hif 发病机制的基因组特征。
Hif KR494 基因组与其他流感嗜血杆菌具有大片段的基因同线性,但也存在明显的基因组重排。预测的 Hif 核心基因组由 1390 个基因组成,与参考株共享,并且揭示了 6 个独特的基因组区域,包含了 191 个独特编码序列的一半。这些区域大部分是插入的遗传片段,很可能来自密切相关的流感嗜血杆菌属,包括埃及嗜血杆菌、溶血嗜血杆菌和副流感嗜血杆菌。重要的是,KR494 基因组具有几个不同于非 f 型菌株的潜在毒力基因。其中包括 sap2 操纵子、aef3 菌毛、卡那霉素核苷转移酶、铁利用蛋白和潜在的 YadA 样三聚体自转运蛋白基因,这些基因可能增加细菌的毒力。此外,Hif KR494 缺乏从头合成组氨酸的 hisABCDEFGH 操纵子、脂寡糖生物合成和生物膜形成的 hmg 基因座、流感嗜血杆菌抗生素抗性岛和流感嗜血杆菌次级钼转运系统。我们通过功能实验证实了 Hif 的组氨酸营养缺陷和卡那霉素耐药性。此外,从不同年份和地理区域获得的 20 株 Hif 临床分离株的 Hif KR494 的独特或缺失基因模式相似。种间比较表明,Hif 基因组与埃及嗜血杆菌的相似性大于 Hid 和 NTHi。
基因组比较分析有助于确定与 Hif 特定毒力相关的基因型特征。与非 f 型流感嗜血杆菌菌株相比,Hif 基因组在参与代谢和存活的成分上存在差异,这可能有助于其侵袭性。