Zobor Ditta, Balousha Ghassan, Baumann Britta, Wissinger Bernd
Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Germany.
Department of Pathology and Histology, Al-Quds University, Eastern Jerusalem, Palestinian Authority.
Mol Vis. 2014 Feb 7;20:178-82. eCollection 2014.
Retinitis pigmentosa (RP) is a heterogenous group of inherited retinal degenerations caused by mutations in at least 45 genes. Recently, the FAM161A gene was identified as the causative gene for RP28, an autosomal recessive form of RP.
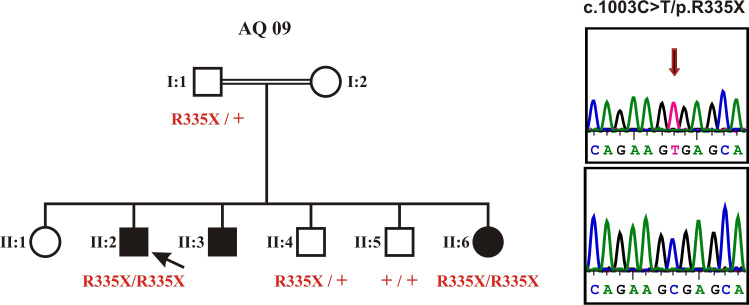
We performed a clinical and molecular genetic study of a consanguineous Palestinian family with two three siblings affected with retinitis pigmentosa. DNA samples were collected from the index patient, his father, his affected sister, and two non-affected brothers. DNA sample from the index was subjected to high resolution genome-wide SNP array. Assuming identity-by-descent in this consanguineous family we applied homozygosity mapping to identify disease causing genes.
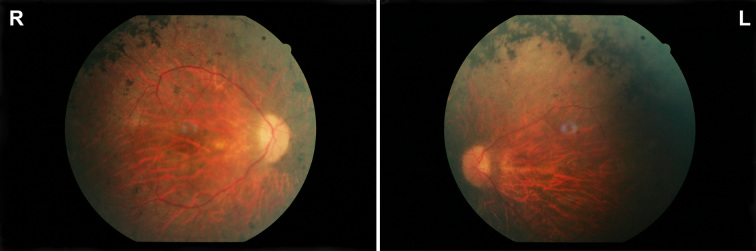
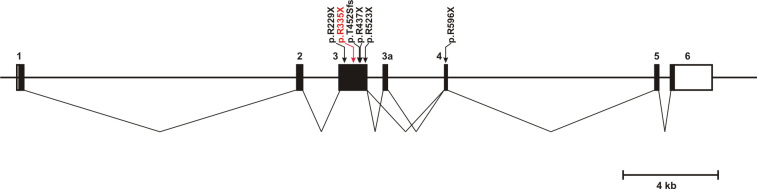
The index patient reported night blindness since the age of 20 years, followed by moderate disease progression with decrease of peripheral vision, the development of photophobia and later on reduced central vision. At the age of 40 his visual acuity was counting fingers (CF) for both eyes, color discrimination was not possible and his visual fields were severely constricted. Funduscopic examination revealed a typical appearance of advanced RP with optic disc pallor, narrowed retinal vessels, bone-spicule like pigmentary changes in the mid-periphery and atrophic changes in the macula. His younger affected brother (37 years) was reported with overall milder symptoms, while the youngest sister (21 years) reported problems only with night vision. Applying high-density SNP arrays we identified several homozygous genomic regions one of which included the recently identified FAM161A gene mutated in RP28-linked autosomal recessive RP. Sequencing analysis revealed the presence of a novel homozygous nonsense mutation, c.1003C>T/p.R335X in the index patient and the affected sister.
We identified an RP28-linked RP family in the Palestinian population caused by a novel nonsense mutation in FAM161A. RP in this family shows a typical disease onset with moderate to rapid progression into severe visual impairment including central vision in the index and overall milder symptoms in the younger brother and sister.
视网膜色素变性(RP)是一组由至少45个基因突变引起的遗传性视网膜变性疾病。最近,FAM161A基因被确定为RP28型视网膜色素变性的致病基因,RP28是一种常染色体隐性遗传形式的视网膜色素变性。
我们对一个近亲结婚的巴勒斯坦家庭进行了临床和分子遗传学研究,该家庭中有三对兄弟姐妹患有视网膜色素变性。从先证者、其父亲、患病的妹妹以及两个未患病的兄弟身上采集了DNA样本。先证者的DNA样本进行了高分辨率全基因组SNP阵列检测。鉴于这个近亲家庭中存在同源性遗传,我们应用纯合性定位来确定致病基因。
先证者自20岁起出现夜盲,随后病情中度进展,周边视力下降,畏光症状出现,后期中心视力下降。40岁时,他双眼视力为手动(CF),无法辨别颜色,视野严重受限。眼底检查显示晚期视网膜色素变性的典型表现,包括视盘苍白、视网膜血管变窄、中周边部骨针样色素改变以及黄斑萎缩性改变。他患病的弟弟(37岁)症状总体较轻,而最小的妹妹(21岁)仅报告有夜盲问题。应用高密度SNP阵列,我们确定了几个纯合基因组区域,其中一个区域包含最近在与RP28相关的常染色体隐性视网膜色素变性中发现的FAM161A基因突变。测序分析显示先证者和患病妹妹中存在一个新的纯合无义突变,即c.1003C>T/p.R335X。
我们在巴勒斯坦人群中确定了一个由FAM161A基因新无义突变引起的与RP28相关的视网膜色素变性家族。该家族中的视网膜色素变性具有典型的发病特征,病情从中度到快速进展为严重视力损害,包括先证者的中心视力受损,而弟弟和妹妹的症状总体较轻。