Kastritis Panagiotis L, Rodrigues João P G L M, Bonvin Alexandre M J J
Bijvoet Center for Biomolecular Research, Faculty of Science/Chemistry, Utrecht University , Utrecht, 3584CH, the Netherlands.
J Chem Inf Model. 2014 Mar 24;54(3):826-36. doi: 10.1021/ci4005332. Epub 2014 Feb 27.
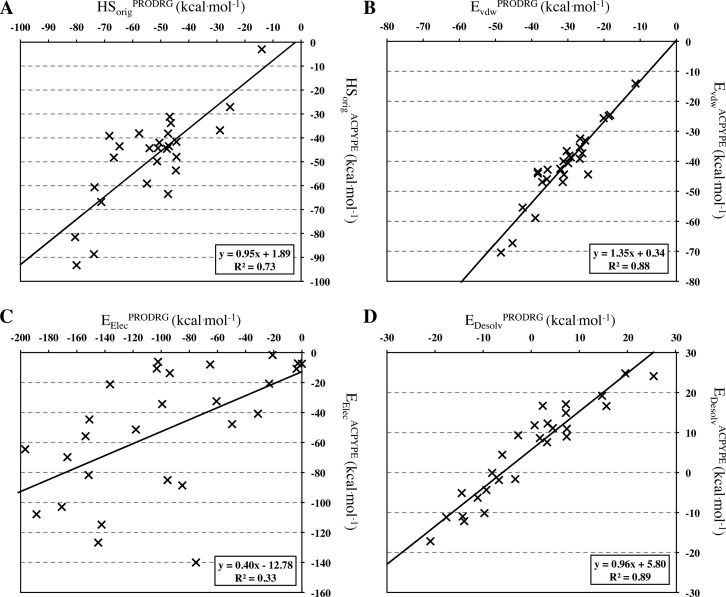
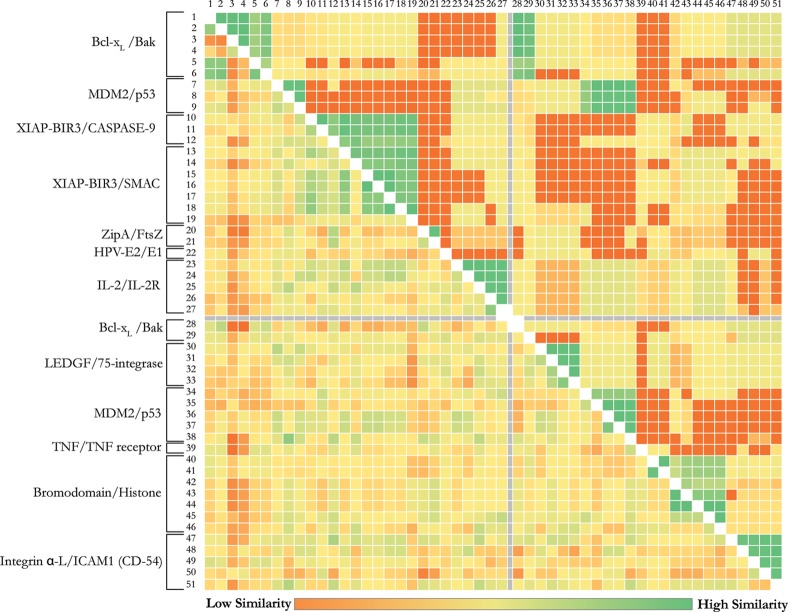
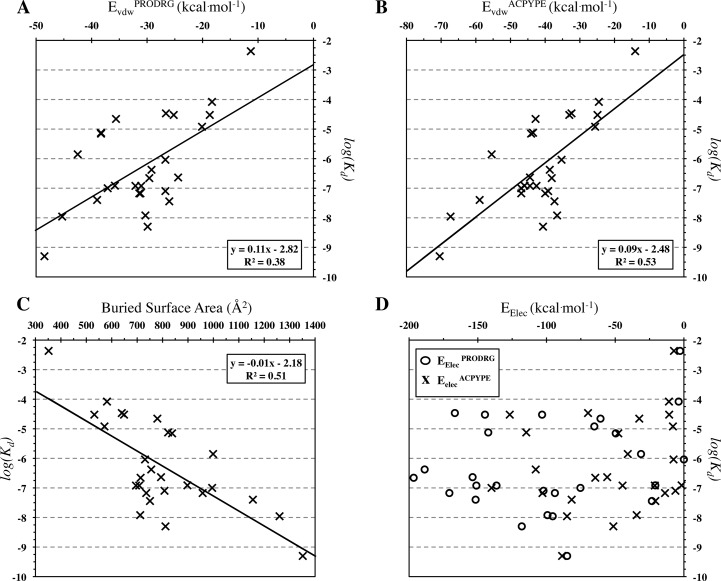
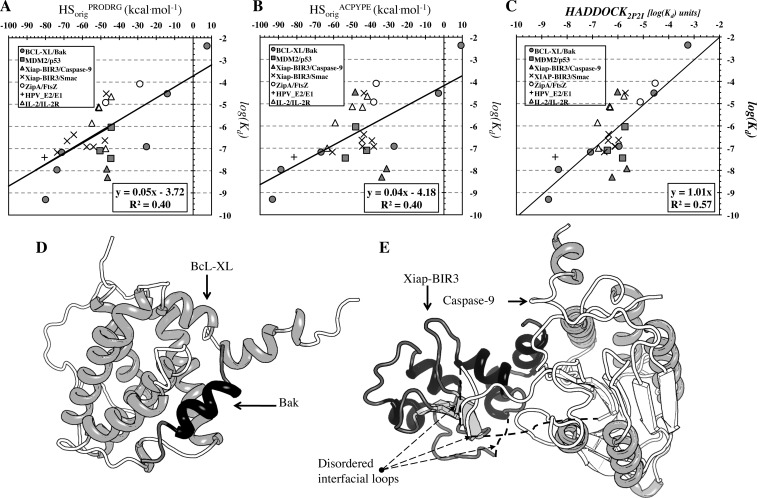
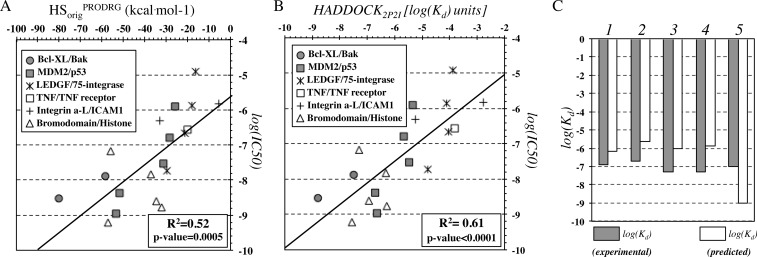
The HADDOCK score, a scoring function for both protein-protein and protein-nucleic acid modeling, has been successful in selecting near-native docking poses in a variety of cases, including those of the CAPRI blind prediction experiment. However, it has yet to be optimized for small molecules, and in particular inhibitors of protein-protein interactions, that constitute an "unmined gold reserve" for drug design ventures. We describe here HADDOCK(2P2I), a biophysical model capable of predicting the binding affinity of protein-protein complex inhibitors close to experimental error (~2-fold larger). The algorithm was trained and 4-fold cross-validated against experimental data for 27 inhibitors targeting 7 protein-protein complexes of various functions and tested on an independent set of 24 different inhibitors for which K(d)/IC50 data are available. In addition, two popular ligand topology generation and parametrization methods (ACPYPE and PRODRG) were assessed. The resulting HADDOCK(2P2I) model, derived from the original HADDOCK score, provides insights into inhibition determinants: while the role of electrostatics and desolvation energies is case-dependent, the interface area plays a more critical role compared to protein-protein interactions.
HADDOCK评分函数用于蛋白质-蛋白质和蛋白质-核酸建模,在多种情况下,包括在CAPRI盲预测实验中,成功地选择了接近天然的对接构象。然而,它尚未针对小分子进行优化,特别是蛋白质-蛋白质相互作用的抑制剂,这些小分子构成了药物设计项目的“未开采金矿”。我们在此描述HADDOCK(2P2I),这是一种生物物理模型,能够预测蛋白质-蛋白质复合物抑制剂的结合亲和力,其预测结果接近实验误差(约大2倍)。该算法针对靶向7种具有不同功能的蛋白质-蛋白质复合物的27种抑制剂的实验数据进行了训练和4倍交叉验证,并在一组独立的24种不同抑制剂上进行了测试,这些抑制剂的K(d)/IC50数据可用。此外,还评估了两种流行的配体拓扑生成和参数化方法(ACPYPE和PRODRG)。从原始HADDOCK评分衍生而来的HADDOCK(2P2I)模型,为抑制决定因素提供了见解:虽然静电和去溶剂化能的作用因情况而异,但与蛋白质-蛋白质相互作用相比,界面面积起着更关键的作用。