dos Santos Marlus Alves, Teixeira Francesco Brugnera, Moreira Heline Hellen Teixeira, Rodrigues Adele Aud, Machado Fabrício Castro, Clemente Tatiana Mordente, Brigido Paula Cristina, Silva Rebecca Tavares e, Purcino Cecílio, Gomes Rafael Gonçalves Barbosa, Bahia Diana, Mortara Renato Arruda, Munte Claudia Elisabeth, Horjales Eduardo, da Silva Claudio Vieira
1] Instituto de Ciências Biomédicas, Universidade Federal de Uberlândia, Uberlândia, MG, Brasil [2].
1] Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos, SP, Brasil [2].
Sci Rep. 2014 Mar 4;4:4259. doi: 10.1038/srep04259.
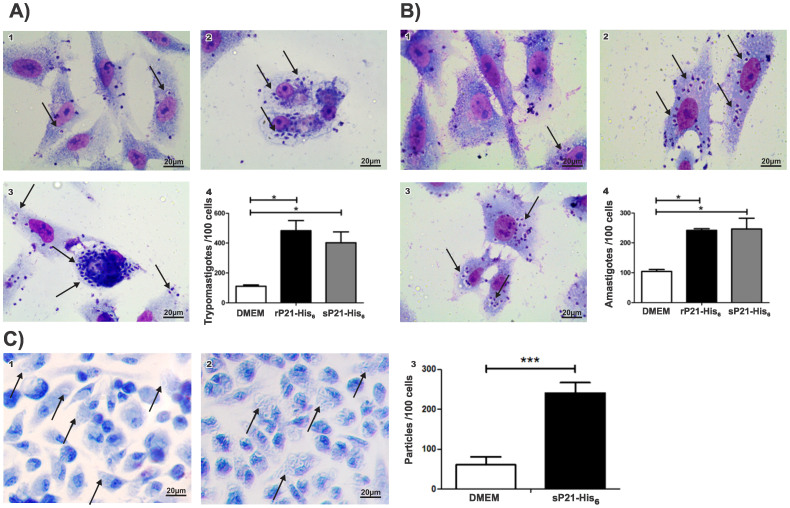
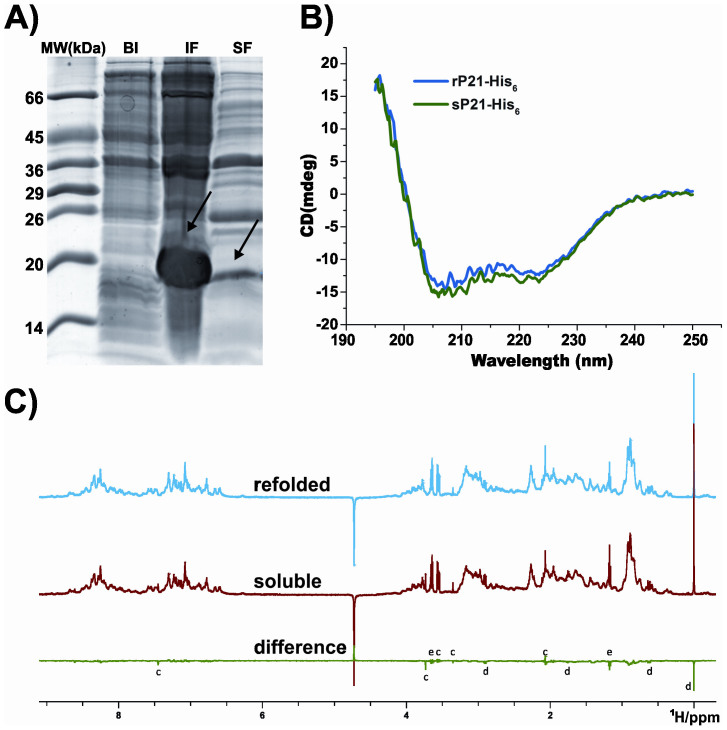
Structural studies of proteins normally require large quantities of pure material that can only be obtained through heterologous expression systems and recombinant technique. In these procedures, large amounts of expressed protein are often found in the insoluble fraction, making protein purification from the soluble fraction inefficient, laborious, and costly. Usually, protein refolding is avoided due to a lack of experimental assays that can validate correct folding and that can compare the conformational population to that of the soluble fraction. Herein, we propose a validation method using simple and rapid 1D (1)H nuclear magnetic resonance (NMR) spectra that can efficiently compare protein samples, including individual information of the environment of each proton in the structure.
蛋白质的结构研究通常需要大量的纯物质,而这些纯物质只能通过异源表达系统和重组技术获得。在这些过程中,大量表达的蛋白质常常存在于不溶性部分,使得从可溶性部分纯化蛋白质效率低下、费力且成本高昂。通常,由于缺乏能够验证正确折叠并能将构象群体与可溶性部分的构象群体进行比较的实验分析方法,蛋白质复性往往被避免。在此,我们提出一种使用简单快速的一维(¹H)核磁共振(NMR)光谱的验证方法,该方法能够有效地比较蛋白质样品,包括结构中每个质子环境的个体信息。