Micale Lucia, Augello Bartolomeo, Maffeo Claudia, Selicorni Angelo, Zucchetti Federica, Fusco Carmela, De Nittis Pasquelena, Pellico Maria Teresa, Mandriani Barbara, Fischetto Rita, Boccone Loredana, Silengo Margherita, Biamino Elisa, Perria Chiara, Sotgiu Stefano, Serra Gigliola, Lapi Elisabetta, Neri Marcella, Ferlini Alessandra, Cavaliere Maria Luigia, Chiurazzi Pietro, Monica Matteo Della, Scarano Gioacchino, Faravelli Francesca, Ferrari Paola, Mazzanti Laura, Pilotta Alba, Patricelli Maria Grazia, Bedeschi Maria Francesca, Benedicenti Francesco, Prontera Paolo, Toschi Benedetta, Salviati Leonardo, Melis Daniela, Di Battista Eliana, Vancini Alessandra, Garavelli Livia, Zelante Leopoldo, Merla Giuseppe
Medical Genetics Unit, IRCCS Casa Sollievo Della Sofferenza Hospital, San Giovanni Rotondo, Italy.
Hum Mutat. 2014 Jul;35(7):841-50. doi: 10.1002/humu.22547. Epub 2014 Apr 9.
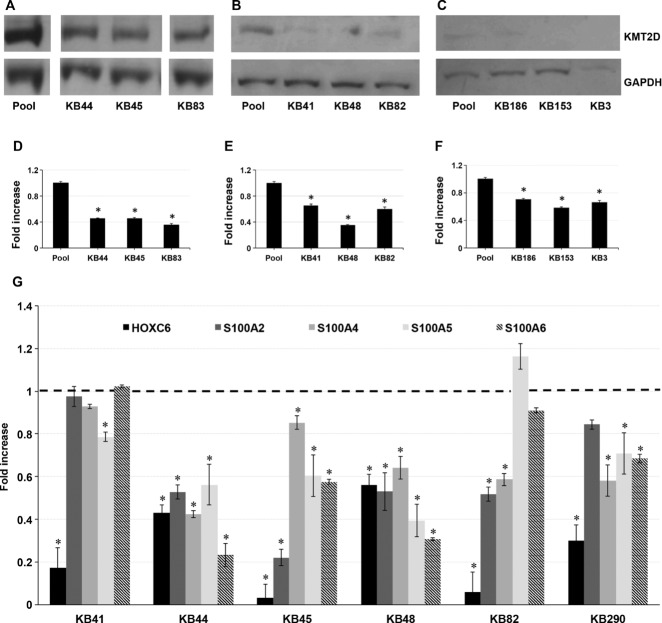
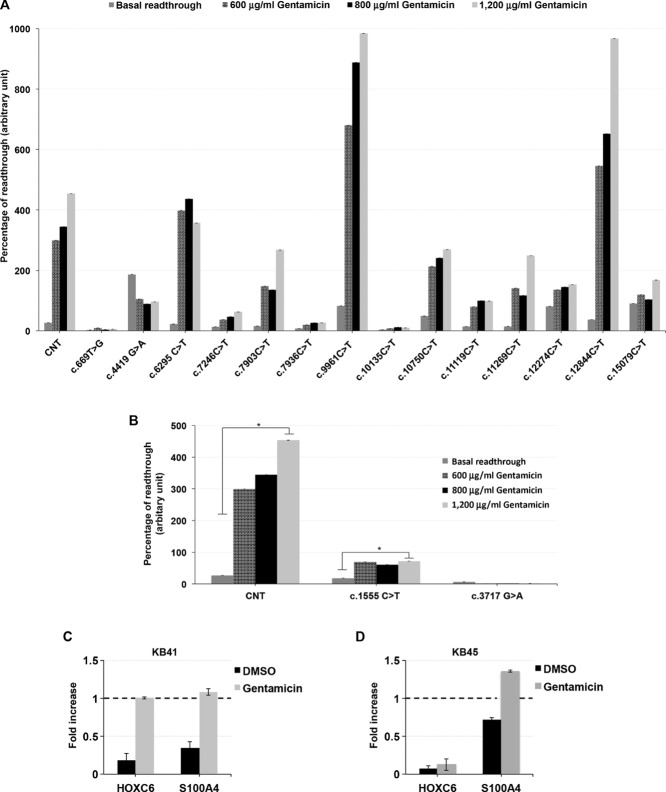
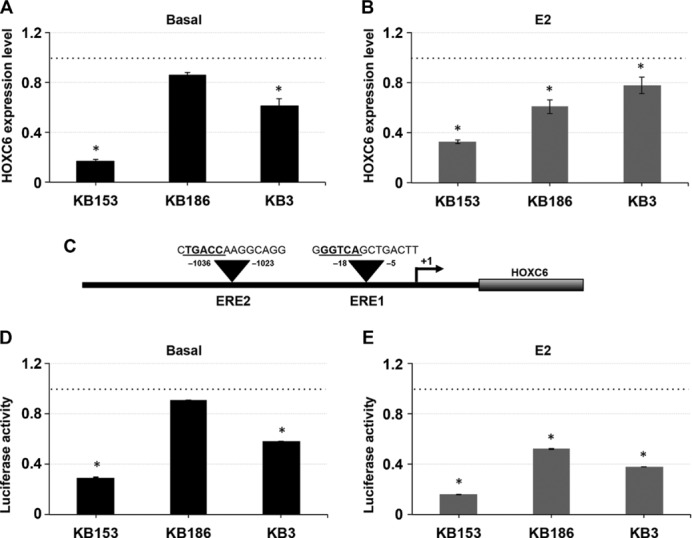
Kabuki syndrome (KS) is a multiple congenital anomalies syndrome characterized by characteristic facial features and varying degrees of mental retardation, caused by mutations in KMT2D/MLL2 and KDM6A/UTX genes. In this study, we performed a mutational screening on 303 Kabuki patients by direct sequencing, MLPA, and quantitative PCR identifying 133 KMT2D, 62 never described before, and four KDM6A mutations, three of them are novel. We found that a number of KMT2D truncating mutations result in mRNA degradation through the nonsense-mediated mRNA decay, contributing to protein haploinsufficiency. Furthermore, we demonstrated that the reduction of KMT2D protein level in patients' lymphoblastoid and skin fibroblast cell lines carrying KMT2D-truncating mutations affects the expression levels of known KMT2D target genes. Finally, we hypothesized that the KS patients may benefit from a readthrough therapy to restore physiological levels of KMT2D and KDM6A proteins. To assess this, we performed a proof-of-principle study on 14 KMT2D and two KDM6A nonsense mutations using specific compounds that mediate translational readthrough and thereby stimulate the re-expression of full-length functional proteins. Our experimental data showed that both KMT2D and KDM6A nonsense mutations displayed high levels of readthrough in response to gentamicin treatment, paving the way to further studies aimed at eventually treating some Kabuki patients with readthrough inducers.
歌舞伎综合征(KS)是一种多发性先天性异常综合征,其特征为独特的面部特征和不同程度的智力迟钝,由KMT2D/MLL2和KDM6A/UTX基因突变引起。在本研究中,我们通过直接测序、多重连接探针扩增(MLPA)和定量PCR对303例歌舞伎综合征患者进行了突变筛查,鉴定出133个KMT2D突变,其中62个此前从未被描述过,以及4个KDM6A突变,其中3个是新发现的。我们发现,一些KMT2D截短突变通过无义介导的mRNA降解导致mRNA降解,导致蛋白质单倍剂量不足。此外,我们证明,在携带KMT2D截短突变的患者淋巴母细胞和皮肤成纤维细胞系中,KMT2D蛋白水平的降低会影响已知KMT2D靶基因的表达水平。最后,我们推测歌舞伎综合征患者可能受益于通读疗法,以恢复KMT2D和KDM6A蛋白的生理水平。为了评估这一点,我们使用介导翻译通读从而刺激全长功能蛋白重新表达的特定化合物,对14个KMT2D和2个KDM6A无义突变进行了原理验证研究。我们的实验数据表明,KMT2D和KDM6A无义突变在庆大霉素治疗下均表现出高水平的通读,为最终用通读诱导剂治疗一些歌舞伎综合征患者的进一步研究铺平了道路。