Duarte Fernanda, Bauer Paul, Barrozo Alexandre, Amrein Beat Anton, Purg Miha, Aqvist Johan, Kamerlin Shina Caroline Lynn
Department of Cell and Molecular Biology, Uppsala University , BMC Box 596, S-751 24 Uppsala, Sweden.
J Phys Chem B. 2014 Apr 24;118(16):4351-62. doi: 10.1021/jp501737x. Epub 2014 Apr 15.
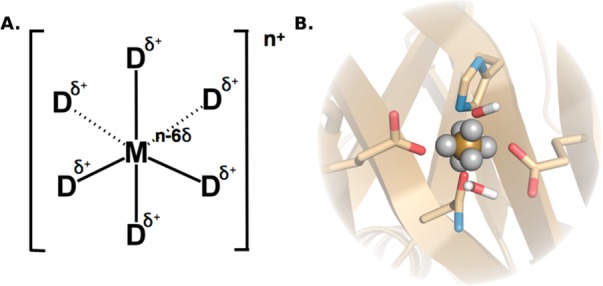
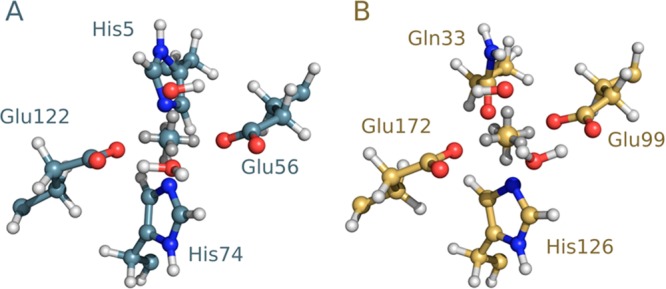
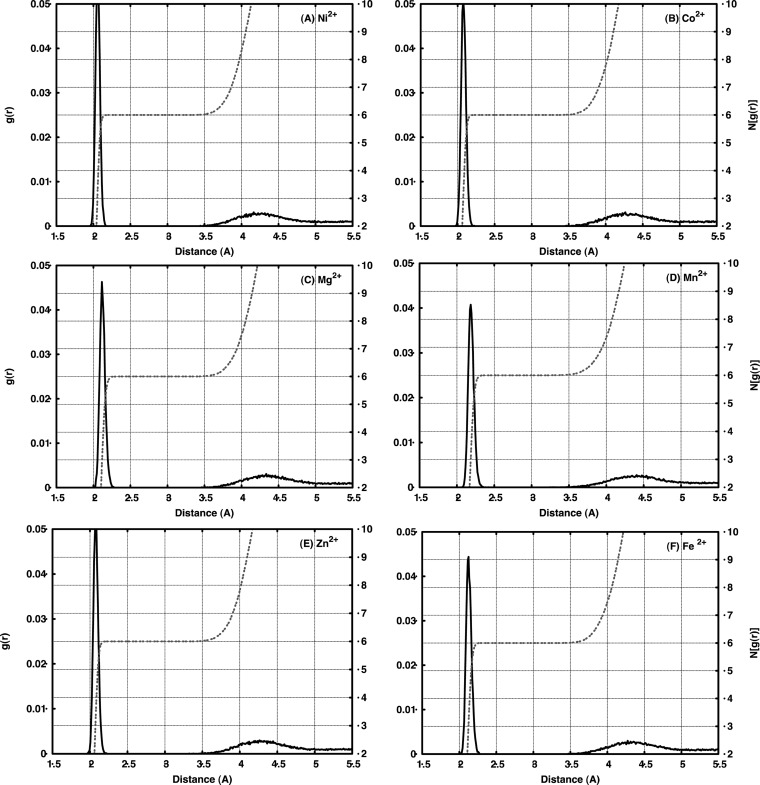
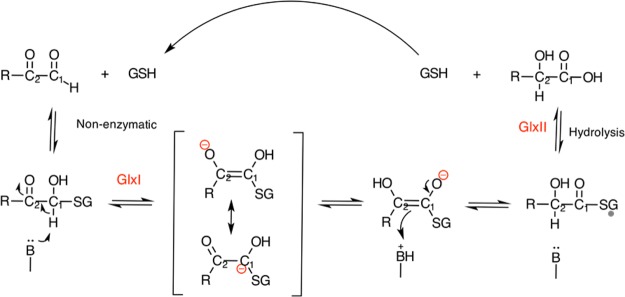
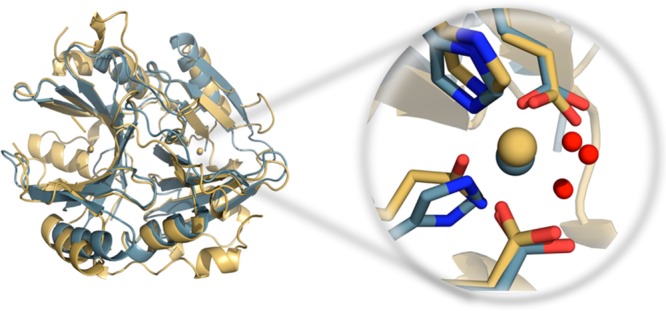
The cationic dummy atom approach provides a powerful nonbonded description for a range of alkaline-earth and transition-metal centers, capturing both structural and electrostatic effects. In this work we refine existing literature parameters for octahedrally coordinated Mn(2+), Zn(2+), Mg(2+), and Ca(2+), as well as providing new parameters for Ni(2+), Co(2+), and Fe(2+). In all the cases, we are able to reproduce both M(2+)-O distances and experimental solvation free energies, which has not been achieved to date for transition metals using any other model. The parameters have also been tested using two different water models and show consistent performance. Therefore, our parameters are easily transferable to any force field that describes nonbonded interactions using Coulomb and Lennard-Jones potentials. Finally, we demonstrate the stability of our parameters in both the human and Escherichia coli variants of the enzyme glyoxalase I as showcase systems, as both enzymes are active with a range of transition metals. The parameters presented in this work provide a valuable resource for the molecular simulation community, as they extend the range of metal ions that can be studied using classical approaches, while also providing a starting point for subsequent parametrization of new metal centers.
阳离子虚拟原子方法为一系列碱土金属和过渡金属中心提供了强大的非键描述,能够捕捉结构和静电效应。在这项工作中,我们对文献中已有的八面体配位的Mn(2+)、Zn(2+)、Mg(2+)和Ca(2+)的参数进行了优化,同时还提供了Ni(2+)、Co(2+)和Fe(2+)的新参数。在所有情况下,我们都能够重现M(2+)-O距离和实验溶剂化自由能,这是目前使用任何其他模型的过渡金属都无法实现的。这些参数还使用了两种不同的水模型进行了测试,表现出一致的性能。因此,我们的参数可以很容易地转移到任何使用库仑和 Lennard-Jones 势描述非键相互作用的力场中。最后,我们以乙二醛酶I的人类和大肠杆菌变体作为展示系统,证明了我们参数的稳定性,因为这两种酶对一系列过渡金属都有活性。这项工作中提出的参数为分子模拟社区提供了宝贵的资源,因为它们扩展了可以使用经典方法研究的金属离子范围,同时也为后续新金属中心的参数化提供了起点。