Stark Amy L, Hause Ronald J, Gorsic Lidija K, Antao Nirav N, Wong Shan S, Chung Sophie H, Gill Daniel F, Im Hae K, Myers Jamie L, White Kevin P, Jones Richard Baker, Dolan M Eileen
Department of Medicine, The University of Chicago, Chicago, Illinois, United States of America.
Committee on Genetics, Genomics and Systems Biology, The University of Chicago, Chicago, Illinois, United States of America; Ben May Department for Cancer Research, The University of Chicago, Chicago, Illinois, United States of America; Institute for Genomics and Systems Biology, The University of Chicago, Chicago, Illinois, United States of America.
PLoS Genet. 2014 Apr 3;10(4):e1004192. doi: 10.1371/journal.pgen.1004192. eCollection 2014 Apr.
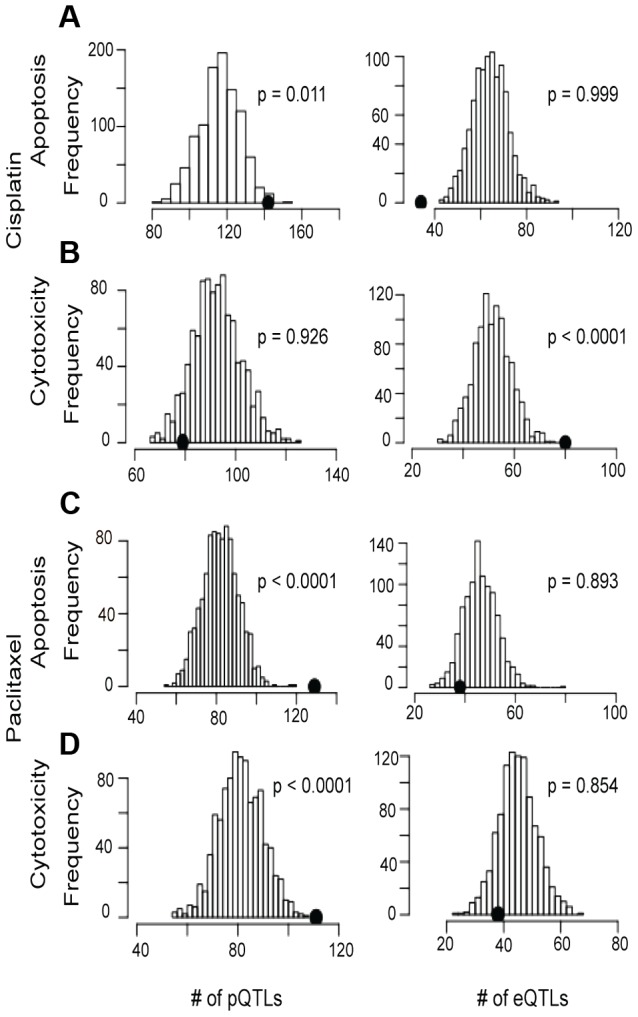
Annotating and interpreting the results of genome-wide association studies (GWAS) remains challenging. Assigning function to genetic variants as expression quantitative trait loci is an expanding and useful approach, but focuses exclusively on mRNA rather than protein levels. Many variants remain without annotation. To address this problem, we measured the steady state abundance of 441 human signaling and transcription factor proteins from 68 Yoruba HapMap lymphoblastoid cell lines to identify novel relationships between inter-individual protein levels, genetic variants, and sensitivity to chemotherapeutic agents. Proteins were measured using micro-western and reverse phase protein arrays from three independent cell line thaws to permit mixed effect modeling of protein biological replicates. We observed enrichment of protein quantitative trait loci (pQTLs) for cellular sensitivity to two commonly used chemotherapeutics: cisplatin and paclitaxel. We functionally validated the target protein of a genome-wide significant trans-pQTL for its relevance in paclitaxel-induced apoptosis. GWAS overlap results of drug-induced apoptosis and cytotoxicity for paclitaxel and cisplatin revealed unique SNPs associated with the pharmacologic traits (at p<0.001). Interestingly, GWAS SNPs from various regions of the genome implicated the same target protein (p<0.0001) that correlated with drug induced cytotoxicity or apoptosis (p ≤ 0.05). Two genes were functionally validated for association with drug response using siRNA: SMC1A with cisplatin response and ZNF569 with paclitaxel response. This work allows pharmacogenomic discovery to progress from the transcriptome to the proteome and offers potential for identification of new therapeutic targets. This approach, linking targeted proteomic data to variation in pharmacologic response, can be generalized to other studies evaluating genotype-phenotype relationships and provide insight into chemotherapeutic mechanisms.
注释和解读全基因组关联研究(GWAS)的结果仍然具有挑战性。将基因变异指定为表达数量性状位点的功能是一种不断扩展且有用的方法,但它仅专注于mRNA而非蛋白质水平。许多变异仍未得到注释。为了解决这个问题,我们测量了来自68个约鲁巴人HapMap淋巴母细胞系的441种人类信号传导和转录因子蛋白的稳态丰度,以确定个体间蛋白质水平、基因变异与化疗药物敏感性之间的新关系。使用微蛋白质免疫印迹法和反相蛋白质阵列从三个独立的细胞系冻融样本中测量蛋白质,以允许对蛋白质生物学重复进行混合效应建模。我们观察到细胞对两种常用化疗药物顺铂和紫杉醇的敏感性存在蛋白质数量性状位点(pQTL)富集。我们在功能上验证了一个全基因组显著的反式pQTL的靶蛋白在紫杉醇诱导的细胞凋亡中的相关性。GWAS对紫杉醇和顺铂药物诱导的细胞凋亡和细胞毒性的重叠结果揭示了与药理学性状相关的独特单核苷酸多态性(p<0.001)。有趣的是,来自基因组各个区域的GWAS单核苷酸多态性涉及与药物诱导的细胞毒性或细胞凋亡相关的同一靶蛋白(p<0.0001)(p≤0.05)。使用小干扰RNA在功能上验证了两个基因与药物反应的关联:SMC1A与顺铂反应相关,ZNF569与紫杉醇反应相关。这项工作使药物基因组学发现从转录组发展到蛋白质组,并为鉴定新的治疗靶点提供了潜力。这种将靶向蛋白质组学数据与药理反应变异联系起来的方法可以推广到其他评估基因型-表型关系的研究中,并为化疗机制提供见解。