Schurch Nicholas J, Cole Christian, Sherstnev Alexander, Song Junfang, Duc Céline, Storey Kate G, McLean W H Irwin, Brown Sara J, Simpson Gordon G, Barton Geoffrey J
Division of Computational Biology, University of Dundee, Dundee, United Kingdom; Division of Biological Chemistry and Drug Discovery, University of Dundee, Dundee, United Kingdom; Centre for Gene Regulation and Expression, University of Dundee, Dundee, United Kingdom.
Division of Computational Biology, University of Dundee, Dundee, United Kingdom.
PLoS One. 2014 Apr 10;9(4):e94270. doi: 10.1371/journal.pone.0094270. eCollection 2014.
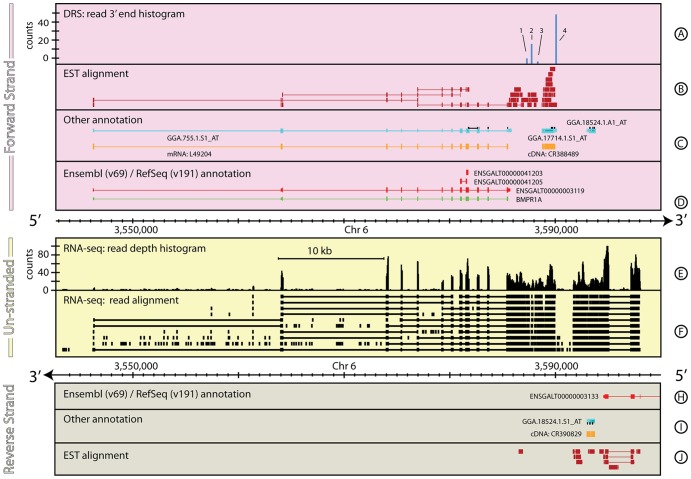
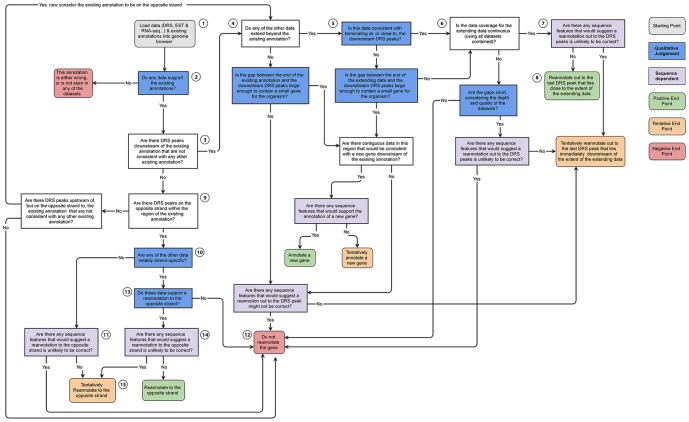
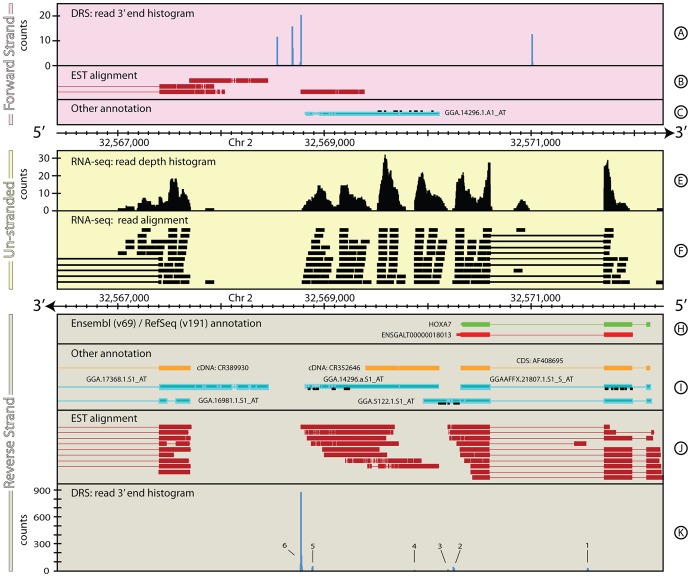
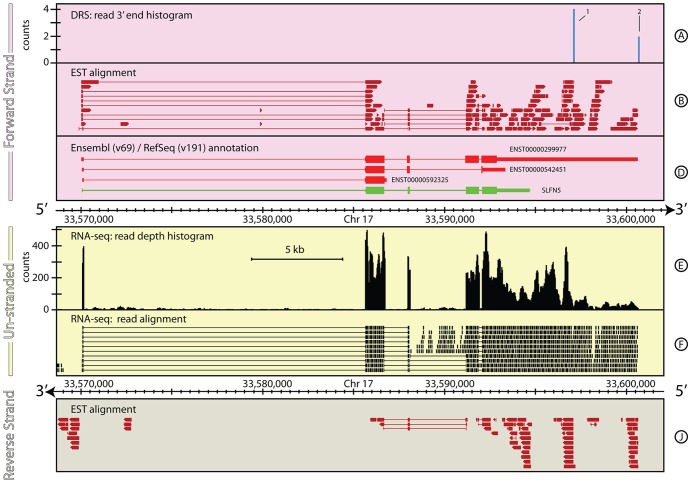
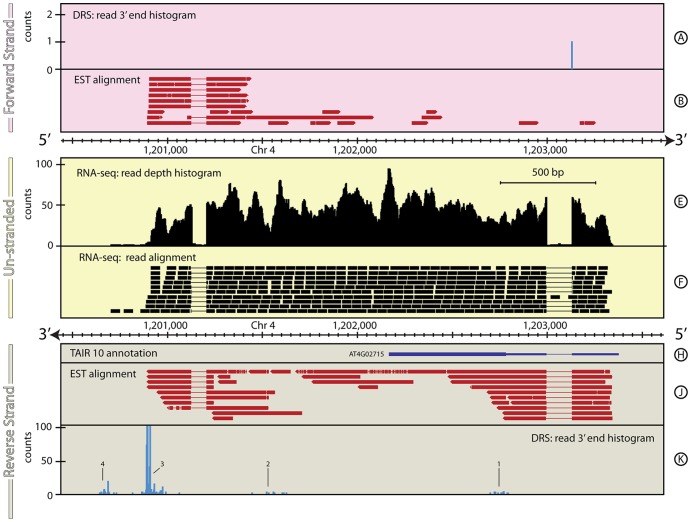
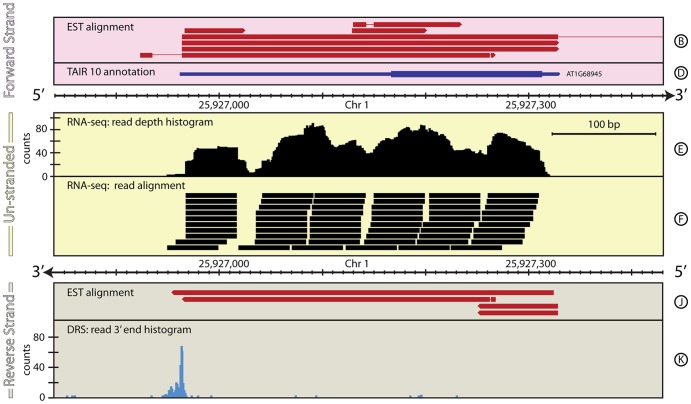
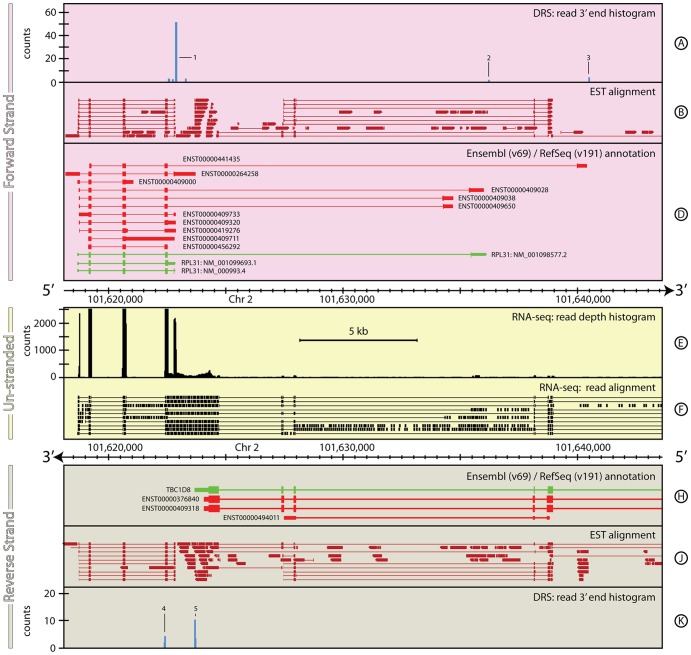
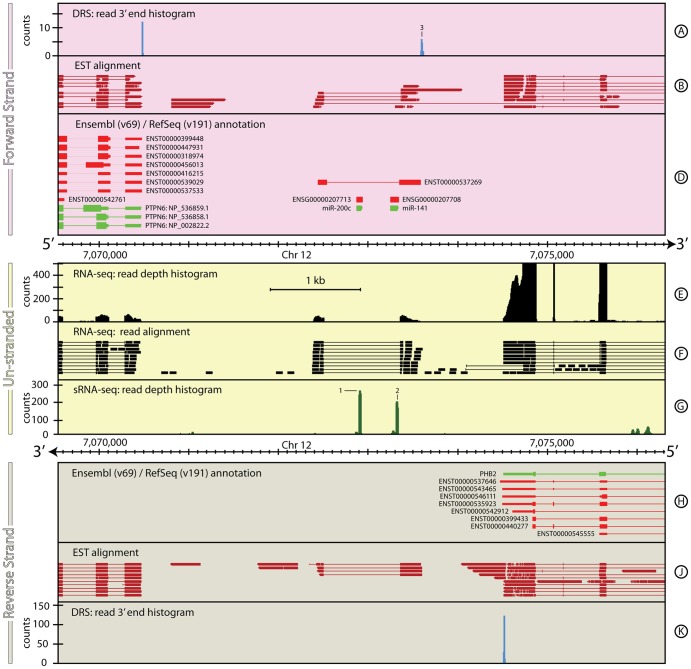
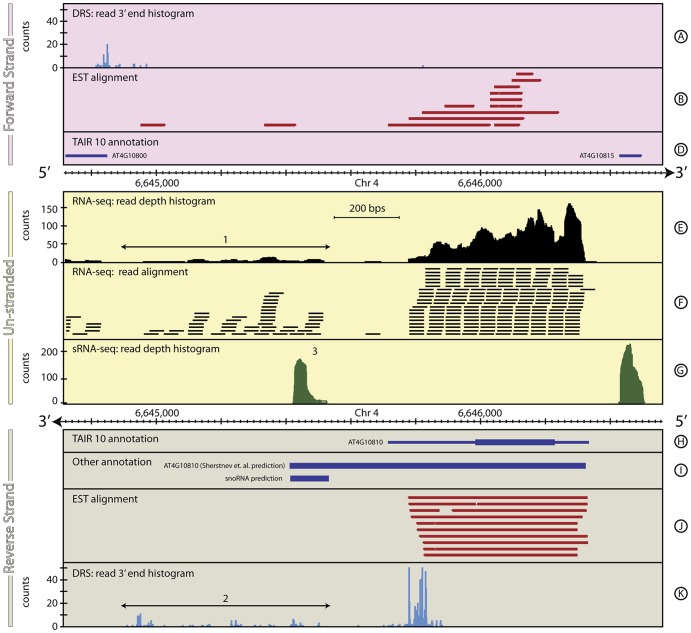
The reference annotations made for a genome sequence provide the framework for all subsequent analyses of the genome. Correct and complete annotation in addition to the underlying genomic sequence is particularly important when interpreting the results of RNA-seq experiments where short sequence reads are mapped against the genome and assigned to genes according to the annotation. Inconsistencies in annotations between the reference and the experimental system can lead to incorrect interpretation of the effect on RNA expression of an experimental treatment or mutation in the system under study. Until recently, the genome-wide annotation of 3' untranslated regions received less attention than coding regions and the delineation of intron/exon boundaries. In this paper, data produced for samples in Human, Chicken and A. thaliana by the novel single-molecule, strand-specific, Direct RNA Sequencing technology from Helicos Biosciences which locates 3' polyadenylation sites to within +/- 2 nt, were combined with archival EST and RNA-Seq data. Nine examples are illustrated where this combination of data allowed: (1) gene and 3' UTR re-annotation (including extension of one 3' UTR by 5.9 kb); (2) disentangling of gene expression in complex regions; (3) clearer interpretation of small RNA expression and (4) identification of novel genes. While the specific examples displayed here may become obsolete as genome sequences and their annotations are refined, the principles laid out in this paper will be of general use both to those annotating genomes and those seeking to interpret existing publically available annotations in the context of their own experimental data.
为基因组序列所做的参考注释为该基因组随后的所有分析提供了框架。除了基础的基因组序列外,正确且完整的注释在解释RNA测序实验结果时尤为重要,在这类实验中,短序列 reads 会与基因组进行比对,并根据注释被分配到各个基因上。参考注释与实验系统之间的不一致可能会导致对所研究系统中实验处理或突变对RNA表达的影响产生错误解读。直到最近,3'非翻译区的全基因组注释受到的关注都少于编码区以及内含子/外显子边界的划定。在本文中,由Helicos Biosciences公司的新型单分子、链特异性直接RNA测序技术所产生的数据(该技术可将3'聚腺苷酸化位点定位在正负2个核苷酸范围内),与存档的EST和RNA测序数据相结合,这些数据来自人类、鸡和拟南芥的样本。文中展示了九个例子,说明这种数据组合能够实现:(1)基因和3'UTR的重新注释(包括将一个3'UTR延长5.9 kb);(2)解析复杂区域中的基因表达;(3)更清晰地解读小RNA表达;以及(4)鉴定新基因。虽然随着基因组序列及其注释的完善,这里展示的具体例子可能会过时,但本文阐述的原则对于那些注释基因组的人以及那些试图在自己的实验数据背景下解读现有公开可用注释的人都将具有普遍用途。