1] Department of Cardiology, Boston Children's Hospital, Boston, Massachusetts, USA. [2].
1] Wyss Institute for Biologically Inspired Engineering, School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts, USA. [2].
Nat Med. 2014 Jun;20(6):616-23. doi: 10.1038/nm.3545. Epub 2014 May 11.
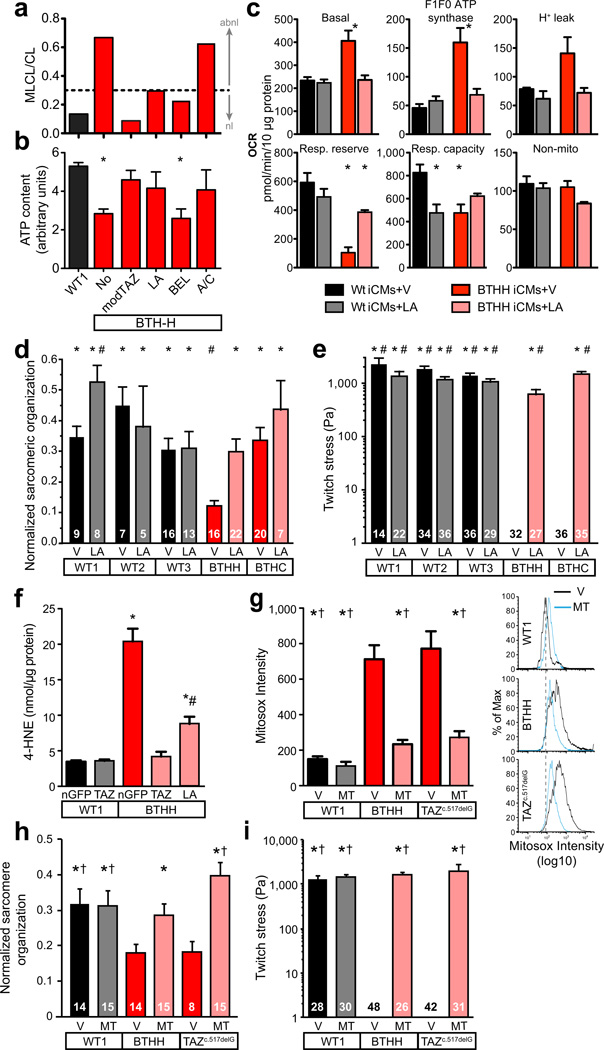
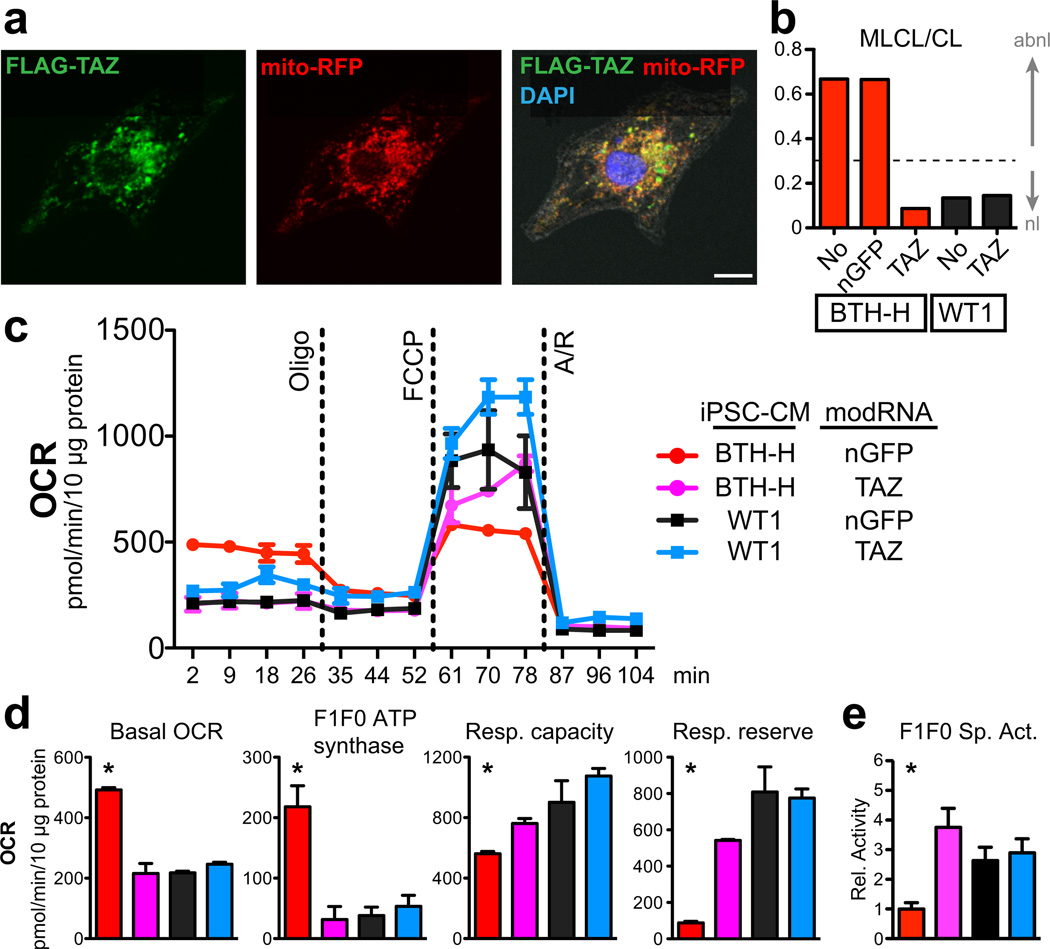
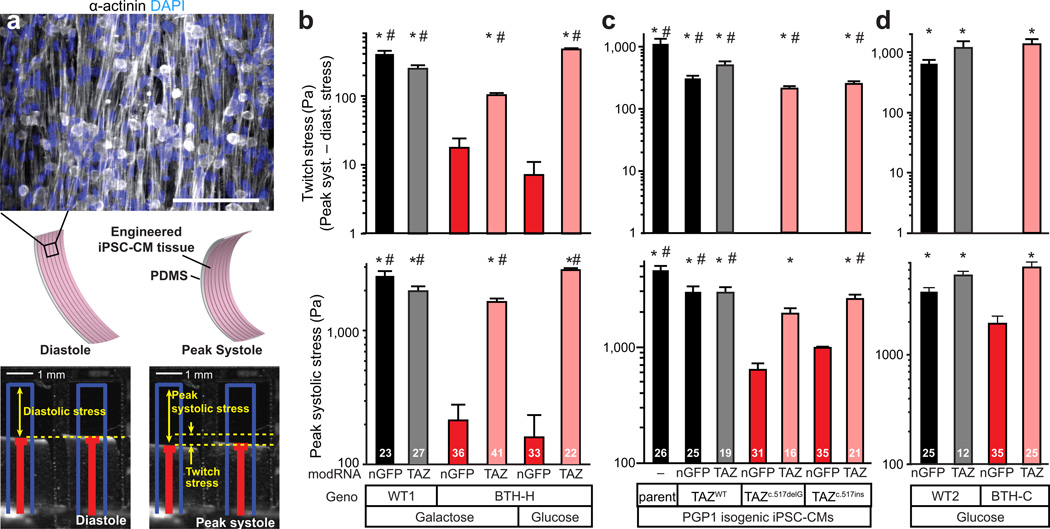
Study of monogenic mitochondrial cardiomyopathies may yield insights into mitochondrial roles in cardiac development and disease. Here, we combined patient-derived and genetically engineered induced pluripotent stem cells (iPSCs) with tissue engineering to elucidate the pathophysiology underlying the cardiomyopathy of Barth syndrome (BTHS), a mitochondrial disorder caused by mutation of the gene encoding tafazzin (TAZ). Using BTHS iPSC-derived cardiomyocytes (iPSC-CMs), we defined metabolic, structural and functional abnormalities associated with TAZ mutation. BTHS iPSC-CMs assembled sparse and irregular sarcomeres, and engineered BTHS 'heart-on-chip' tissues contracted weakly. Gene replacement and genome editing demonstrated that TAZ mutation is necessary and sufficient for these phenotypes. Sarcomere assembly and myocardial contraction abnormalities occurred in the context of normal whole-cell ATP levels. Excess levels of reactive oxygen species mechanistically linked TAZ mutation to impaired cardiomyocyte function. Our study provides new insights into the pathogenesis of Barth syndrome, suggests new treatment strategies and advances iPSC-based in vitro modeling of cardiomyopathy.
研究单基因线粒体心肌病可能深入了解线粒体在心脏发育和疾病中的作用。在这里,我们结合患者来源的和基因工程诱导多能干细胞(iPSC)与组织工程学,阐明了由编码 tafazzin(TAZ)的基因突变引起的线粒体疾病——Barth 综合征(BTHS)的心肌病的病理生理学基础。使用 BTHS iPSC 衍生的心肌细胞(iPSC-CMs),我们定义了与 TAZ 突变相关的代谢、结构和功能异常。BTHS iPSC-CMs 组装稀疏且不规则的肌节,并且工程化的 BTHS“芯片上心脏”组织收缩较弱。基因替换和基因组编辑表明 TAZ 突变对于这些表型是必需和充分的。在整个细胞 ATP 水平正常的情况下,肌节组装和心肌收缩异常发生。过量的活性氧物质将 TAZ 突变与受损的心肌细胞功能机械地联系起来。我们的研究为 Barth 综合征的发病机制提供了新的见解,为治疗策略提供了新的建议,并推进了基于 iPSC 的心肌病体外建模。